





| International Journal of Clinical Pediatrics, ISSN 1927-1255 print, 1927-1263 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, Int J Clin Pediatr and Elmer Press Inc |
| Journal website http://www.theijcp.org |
Case Report
Volume 2, Number 1, June 2013, pages 44-47
Griscelli Syndrome Type 2 With Hemophagocytic Lymphohistocytosis a Rare and Lethal Disorder
Kesh Ram Meenaa, b, Pradeep Kumara, Anitaa, Premila Paula
aDepartment of Pediatrics, Vardhman Mahavir Medical College and Safdarjang Hospital, New Delhi, India
bCorresponding author: Kesh Ram Meena, A-21, Mount Everest Apartment, Plot17, Sector 9, Dwarka, New Delhi, India
Manuscript accepted for publication May 14, 2013
Short title: Griscelli Syndrome Type 2
doi: https://doi.org/10.4021/ijcp100w
| Abstract | ▴Top |
A two and half years old male child was admitted with silvery grey hair, recurrent chest infection, ear discharge, hepatosplenomegaly, pancytopenia and hemophagocytosis in bone marrow. Light microscopy of hair shaft showed large unevenly distributed melanin pigment. Skin biopsy showed large coarse aggregates of melanin and the basal layer of epidermis showed reactivity with Masson’s fontana stain. Griscelli syndrome type 2 was made as diagnosis which was confirmed by mutation study which showed nonsense mutation of Exon 6 of RAB 27A gene. The child was in accelerated phase and succumbed to death.
Keywords: Griscelli syndrome; Hemophagocytosis; Nonsense mutation; RAB 27A gene
| Introduction | ▴Top |
Griscelli syndrome is a rare autosomal recessive disorder characterized by pigmentary dilution of hairs, skin and variable cellular immunodeficiency [1]. It was first described by Griscelli and Pruniera in 1978 [2]. Common age of diagnosis is between 4 months to 7 years of age [1]. It is classified into three types. Pigmentary dilution is common in all subtypes. Type 1 is caused by mutation in the MYO5A gene which encodes the motor protein myosin-Va which is highly expressed in the brain and plays a very important role in neurons [3, 4]. Mutation in MYO5A gene is associated with severe primary neurological impairment such as developmental delay and mental retardation [5]. Griscelli syndrome type 2 is caused by mutation in RAB 27A gene. RAB 27A gene encodes the small GTPase protein Rab27a. Rab27a is associated with melanosomes in pigment cells and regulates melanosome transport via its interaction with actin based cellular motors such as myosin-Va and myosin V11a. Myosin-Va and myosin V11a do not interact directly with Rab27a but require linker proteins, melanophilin and myrip respectively. RAB27A gene mutation results in pigmentary dilution of skin and hair, the presence of large clumps of pigment in hair shafts, and an accumulation of melanosomes in melanocytes. Rab27a is also required for secretion of lytic granules in cytotoxic T lymphocytes. In RAB27A gene mutation, capacity of the lymphocytes and NK cells to lyse target cells is impaired or absent which results in primary immunodeficiency. Griscelli syndrome type 2 patients also develop an uncontrolled T-lymphocyte and macrophage activation syndrome, known as hemophagocytic lymhohistiocytosis (HLH). Hemophagocytic lymphohistocytosis has been characterized by an unremitting polyclonal CD8+ T cell expansion, and lymphocytic infiltration of visceral tissues that leads to macrophage activation (hemophagocytosis) and release of several cytokines, including interferon gamma, interleukins (IL-1, IL-6, IL-18) and tumor necrosis factor-α, which sustains the macrophage activation and leads to hyperinflammation, involving progressive deterioration of the central nervous system, and multiple organ failure and death without bone marrow transplantation [6]. We report a case of type 2 Griscelli syndrome with hemophagocytic lymphohistiocytosis
| Case Report | ▴Top |
A two year and half years old male child which was born of non-consanguineous marriage was referred to our hospital with complains of fever and cough (1 year, off & on), progressive abdomen distension (3 months), black coloured stool (2 months), and ear discharge (1 month). There were no complains of vomiting, loose stools, burning maturation, jaundice, anasarca, itching, repeated boils, convulsion. There was history of three blood transfusions in last one year. In family there was history of death of eldest female sibling at 9 months of age after having recurrent episode of fever, cough and ear discharge. Hairs of the child were light coloured. On examination he was febrile, his vital parameters were normal. Hairs on scalp, eye brow, eye lashes were silvery grey (Fig. 1). He was having marked pallor with left ear purulent discharge. He had delayed gross motor, fine motor, social and linguistic mile stones. Abdomen was distended with liver 6 cm below costal margin with smooth surface, firm and non-tender in consistency with sharp margins. Liver span was 12 cm. The spleen was palpable 12 cm below left costal margin and was firm in consistency. The skin was of light colour. Rest of the examinations were normal. His investigation revealed haemoglobin 5 g/dL, TLC 3,000/mm3; platelet count 25,000/mm3 and reticulocytes were 7.2%. Peripheral smear showed pancytopenia with normocytic normochromic anaemia. There were no giant cytoplasmic granules in leucocytes, serum bilirubin was 0.7 mg/dL, total protein was 6.8 g/dL and SGPT was 30 IU/L, SGOT 50 IU/L, HIV, HBsAg, HCV, Monteux test and RK-39 were negative. Serum ceruloplasmin was 36 µg/dL (22 - 58 µg/dL), LDH 220 U/L (150 - 500 U/L), coagulation profile was normal. Fibrinogen level was 1.7 g/dL (normal 2 - 4 g/dL), serum ferritin level was 4,000 ng/mL (normal range 6 - 140 ng/mL). Immunological test showed IgG 1,400 mg/dL (350 - 1,180 mg/dL), IgA 140 mg/dL (36 - 165 mg/dL), IgM 80 mg/dL (36 - 160 mg/dL). CSF biochemistry was normal and cytology showed 50 cell (lymphocyte 100%).Chest X-ray was normal. Ultrasonography abdomen showed hepatosplenomegaly with fatty liver. Blood culture showed growth of Acinobacter and urine culture showed growth of candida species. MRI brain showed hyper-intensity in cerebellar hemisphere, Right parital and left temporal lobe, which was suggestive of lympho-histocytic infiltrations (Fig. 2). Bone marrow examination showed erythrophagocytosis. These findings were consistent with Griscelli syndrome type 2. For confirmation of this hair shaft microscopy was done which shown large unevenly distributed melanin pigment. Skin biopsy showed large coarse aggregates of melanin and the basal layer of epidermis showed reactivity with Massion’s Fontana stain. Mutation study was done which showed non-sense mutation of Exon6 of RAB-27A gene. This mutation is the cause of Griscelli syndrome. The child was treated with IV antibiotics, antifungal medicine and packed cell transfusion. Initially he responded well but after two weeks he showed neurological deterioration for which Methyl prednisolone was given after which he was improved. He was discharged and planned for Bone marrow transplantation and HLA matching was done with his elder sibling. But after two weeks he developed severe anaemia and was given packed red cell transfusion, IV antibiotics. However, he continued to deteriorate neurologically and despite repeated course of methyl prednisolone, he succumbed to death.
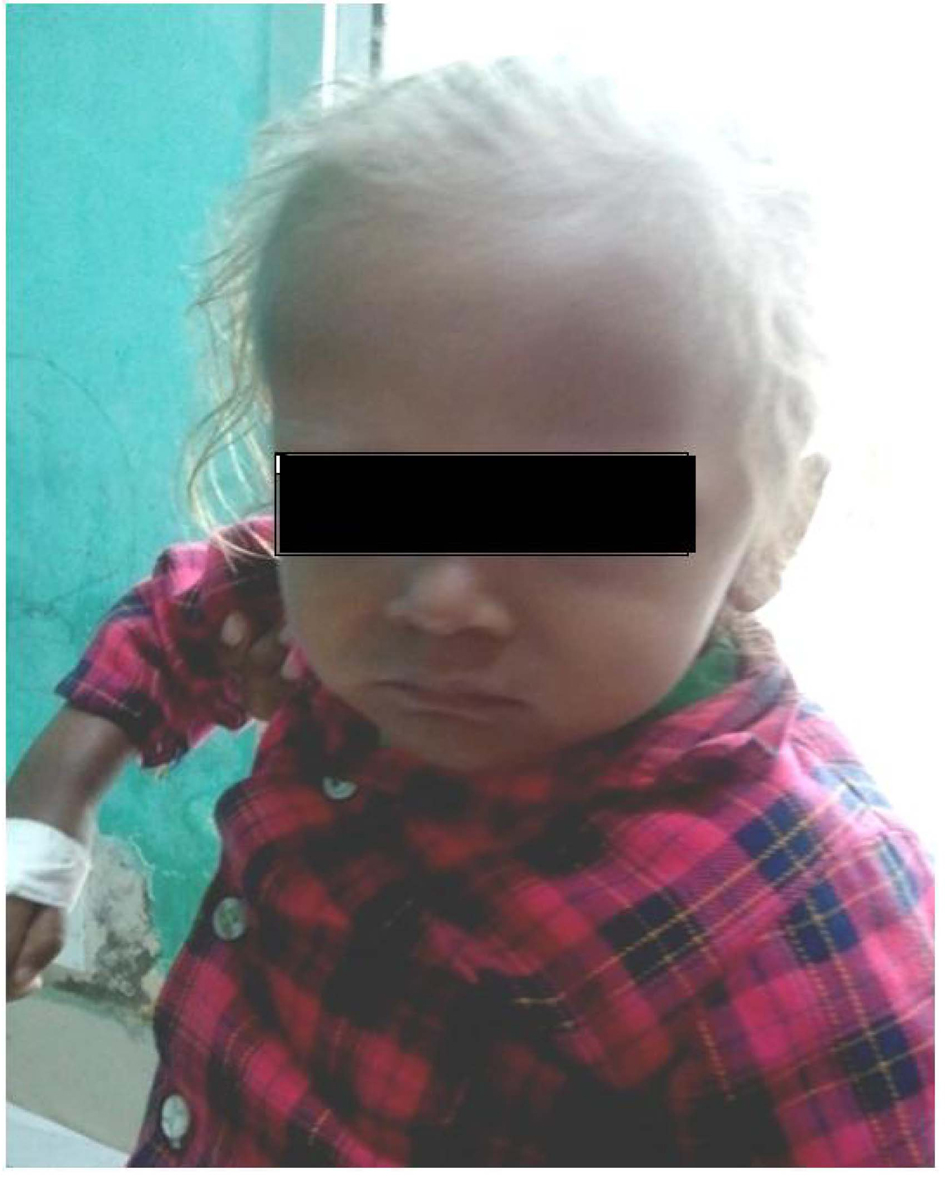 Click for large image | Figure 1. Physical appearance of patient showing silvery grey hair and light colored skin. |
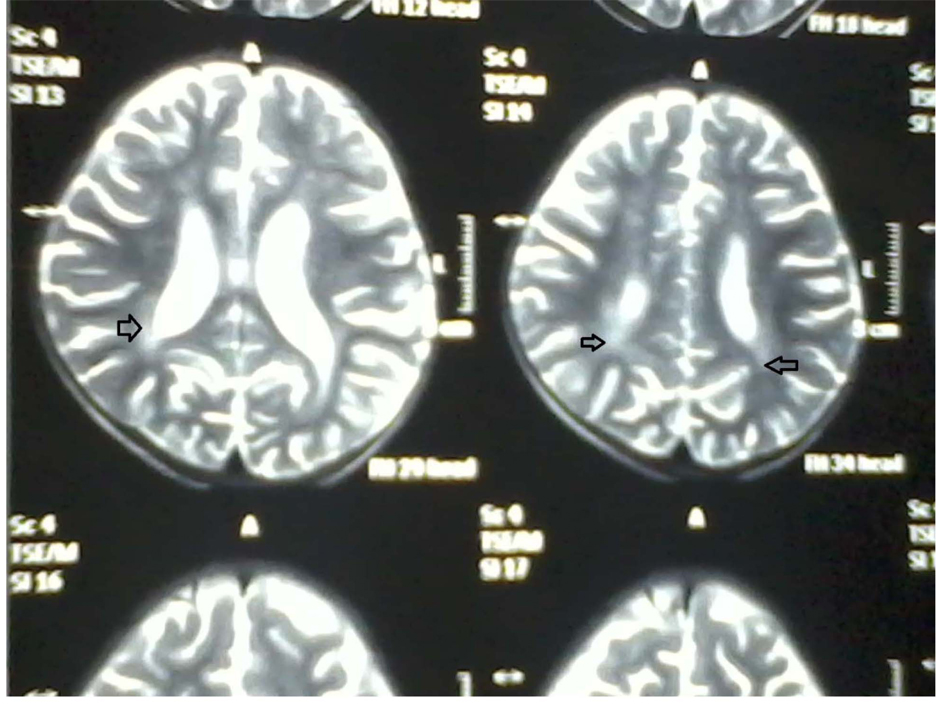 Click for large image | Figure 2. MRI brain showing increased intensity (arrow). |
| Discussion | ▴Top |
Griscelli syndrome is a rare and not widely known disease and its clinical feature such as silvery gray hair and immunological dysfunction resemble Chediak Higashi Syndrome (CHS), an erythrophagocytic lymphohistiocytosis [7]. However, light microscopic examination of scalp hair is an easy way to differentiate these two syndromes. Hair shaft from Chediak Higashi syndrome (CHS) patients reveals small regular melanin aggregates [8], whereas in Griscelli syndrome, large clumps of pigments irregularly distributed are found. The hallmark of CHS is the presence of giant intracytoplasmic granules in virtually all granulocytic cells which is never seen in Griscelli Syndrome. Skin biopsy from CHS patients reveals giant melanosomes in both melanocyte and Keratinocyte while in Griscelli Syndrome melanocyte shows massive accumulation of mature melanosomes with adjacent Keratinocyte containing only sparse melanosomes.
The prognosis and treatment of Griscelli Syndrome depend on the type of the condition. There is no cure for Griscelli Syndrome type I and the life of the patients depends on the severity of neurological impairment. For Griscelli Syndrome type 2 the treatment of choice is bone marrow transplantation [9], which yields better result when done early. Here is an example of how important it is to prepare the health care services to early recognise of this disease and propose effective treatment in time. Cases of successful bone marrow transplantation are reported and immunosuppressive therapy (high dose systemic methylprednisolone, cyclosporine, etoposide) is also used to maintain the patient stable as a palliative therapy or to induce remission until bone marrow transplantation [10, 11]. Bizario et al [12] showed rescuing of cytotoxic T lymphocyte activity using a retroviral vector to mediate the transfer of RAB27A gene, opening an alternative possibility for Griscelli syndrome treatment. Griscelli Syndrome type3 does not need treatment since it involves altered pigmentation only.
Amniotic fluid cells and chorionic villi cells can help in prenatal diagnosis of Griscelli syndrome. Prenatal diagnosis of Griscelli syndrome has been accomplished by examination of hair from a biopsy sample of fetal scalp obtained at 21 weeks of gestation. A fetus that had such a biopsy was aborted. These results were confirmed by a postabortation examination of the fetus revealing silvery hair and characteristic microscopic findings [13].
With cloning of the Griscelli syndrome genes, direct mutation based carrier detection and prenatal diagnosis currently appears possible in families with defined MYOVA or RAB27A gene mutation. In addition, given the proximity of the 2 genes responsible for Griscelli syndrome, polymorphic markers linked to the Griscelli syndrome locus in the band 15q21 region can be used for identifying the presence of the gene even if the precise mutation has not yet been identified in a family [13].
Conclusion
Griscelli syndrome should always be suspected in any child with silvery hair because early bone marrow transplant might offer a chance for survival.
| References | ▴Top |
- Mancini AJ, Chan LS, Paller AS. Partial albinism with immunodeficiency: Griscelli syndrome: report of a case and review of the literature. J Am Acad Dermatol. 1998;38(2 Pt 2):295-300.
doi - Griscelli syndrome,type1:OMIM(on line Mendelian inheritance in man).
- Krendel M, Mooseker MS. Myosins: tails (and heads) of functional diversity. Physiology (Bethesda). 2005;20:239-251.
- Larson RE. Myosin-V: a class of unconventional molecular motors. Braz J Med Biol Res. 1996;29(3):309-318.
pubmed - Pastural E, Barrat FJ, Dufourcq-Lagelouse R, Certain S, Sanal O, Jabado N, Seger R, et al. Griscelli disease maps to chromosome 15q21 and is associated with mutations in the myosin-Va gene. Nat Genet. 1997;16(3):289-292.
doi pubmed - Janka G, Zur Staud U. Familial and acquired hemophagocytic lymphohistocytosis. Hematol Am Soc Educ Program 2005;82-88.
pubmed - Barak Y, Nir E. Chediak-Higashi syndrome. Am J Pediatr Hematol Oncol. 1987;9(1):42-55.
doi - Malhotra AK, Bhaskar G, Nanda M, Kabra M, Singh MK, Ramam M. Griscelli syndrome. J Am Acad Dermatol. 2006;55(2):337-340.
doi pubmed - Klein C, Philippe N, Le Deist F, Fraitag S, Prost C, Durandy A, Fischer A, et al. Partial albinism with immunodeficiency (Griscelli syndrome). J Pediatr. 1994;125(6 Pt 1):886-895.
doi - Aslan D, Sari S, Derinoz O, Dalgic B. Griscelli syndrome: description of a case with Rab27A mutation. Pediatr Hematol Oncol. 2006;23(3):255-261.
doi pubmed - Schuster F, Stachel DK, Schmid I, Baumeister FA, Graubner UB, Weiss M, Haas RJ, et al. Griscelli syndrome: report of the first peripheral blood stem cell transplant and the role of mutations in the RAB27A gene as an indication for BMT. Bone Marrow Transplant. 2001;28(4):409-412.
doi pubmed - Bizario JC, Feldmann J, Castro FA, Menasche G, Jacob CM, Cristofani L, Casella EB, et al. Griscelli syndrome: characterization of a new mutation and rescue of T-cytotoxic activity by retroviral transfer of RAB27A gene. J Clin Immunol. 2004;24(4):397-410.
doi pubmed - Noah S Scheinfeld, An M Johnson et al. Griscelli syndrome work up. eMedicine.Medscape refrence 15 feb.2013.
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
International Journal of Clinical Pediatrics is published by Elmer Press Inc.