





| International Journal of Clinical Pediatrics, ISSN 1927-1255 print, 1927-1263 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, Int J Clin Pediatr and Elmer Press Inc |
| Journal website http://www.theijcp.org |
Case Report
Volume 7, Number 3, September 2018, pages 39-42
Autoimmune Pancreatitis in Two Adolescent Boys
Samer Saadea, d, Cesar Yaghib, Joseph Bou Jaoudeb, Bassam Eidc
aDepartment of Pediatrics, Hotel-Dieu de France Hospital, Beirut, Lebanon
bDepartment of Gastroenterology and Hepatology, Hotel-Dieu de France Hospital, Beirut, Lebanon
cPediatric Gastroenterology and Hepatology Unit, Department of Pediatrics, Hotel-Dieu de France Hospital, Beirut, Lebanon
dCorresponding Author: Samer Saade, Hotel-Dieu de France, Achrafieh, Beirut, Lebanon
Manuscript submitted May 16, 2018, accepted June 13, 2018
Short title: Autoimmune Pancreatitis in Two Cases
doi: https://doi.org/10.14740/ijcp303w
| Abstract | ▴Top |
In this current review, we report the cases of autoimmune pancreatitis in two adolescent boys. The diagnosis was made essentially on the basis of imaging result and response to steroid therapy. One of them developed ulcerative colitis 1 year later, which was in favor of the autoimmune pancreatitis diagnosis. Since the presentation of autoimmune pancreatitis can mimic other conditions, a prompt diagnosis should be made, helping the patient receive adequate treatment and avoid unnecessary surgical interventions.
Keywords: Pancreatitis; Autoimmune; Steroid; Adolescent
| Introduction | ▴Top |
Autoimmune pancreatitis (AIP) is a rare disease that is extremely uncommon in children [1-3]. It represents 2% of chronic pancreatitis occurring generally in adult males [4]. The usual presentation is a pancreatic pseudo-tumor that can often be mistaken for pancreatic cancer. AIP can be divided into two types: 1) Type 1: more common in elderly Asian males, involves other organs and is associated with an increased IgG4 level; 2) Type 2 has been described in younger individuals with normal serum IgG4 levels and is more commonly associated with the development of ulcerative colitis [5]. The International Consensus Diagnostic Criteria (ICDC) included five fundamental criteria for AIP, namely, imaging of pancreatic parenchyma and duct, serology, other organ involvement, pancreatic histology, and an optional criterion of response to steroid therapy [6]. We are presenting the cases of two adolescent boys diagnosed with AIP. Our main objective is raising the awareness about this rare childhood disease to help clinicians having a high index of suspicion for it when present, and providing appropriate diagnostic tools and treatments.
| Case Reports | ▴Top |
Case 1
A 13-year-old Lebanese boy, with an unremarkable previous medical history, presented to the emergency department with a 4-day history of colicky periumbilical pain and nausea. The clinical exam showed no abnormalities, in particular for abdominal and neurological exam.
He was diagnosed with acute pancreatitis based on elevated lipase 7,538 IU/L, amylase 373 IU/L and slightly increased C-reactive protein. Complete blood count (CBC) and liver function tests results were the following: hemoglobin 14 g/dL, WBC 12,600/mm3, platelets 293,000/mm3, ASAT 29 IU, ALAT 50 IU and GGT 74 IU. Creatinine level was 50 μmol/L, while levels of calcium, cholesterol and triglycerides were normal. Abdominal ultrasound demonstrated a dilation of intrahepatic and extrahepatic biliary ducts. Magnetic resonance cholangiopancreatography (MRCP) (Fig. 1) showed irregular aspect and dilation of intrahepatic and extrahepatic biliary ducts with an irregular main pancreatic duct and a bulky, hypertrophied pancreas showing high T2 signal and low T1 signal surrounding the body and the tail. Endoscopic ultrasound showed a heterogeneous pseudo-tumoral parenchyma, compression of the extrahepatic biliary duct with biliary sludge, and a narrow irregular main pancreatic duct. AIP was suspected and workup for autoimmune diseases was undergone, showing the absence of autoantibodies (antinuclear antibodies, rheumatoid factor, smooth muscle antibodies, LKM1 antibodies and mitochondrial antibodies). The level of IgG4 antibodies was at 45.6 mg/dL (reference range between 4 and 230 mg/dL). Multiple serologies done were negative (Epstein-Barr virus, cytomegalovirus, mumps, measles, mycoplasma, hepatitis A, B and C). He was kept nil per os (NPO) for 1 week with total parenteral nutrition resulting in a decrease of lipase level. Enteral feeding was resumed later uneventfully. Control abdominal ultrasound showed regression of the pancreatic size and the patient was later discharged at home symptoms free.
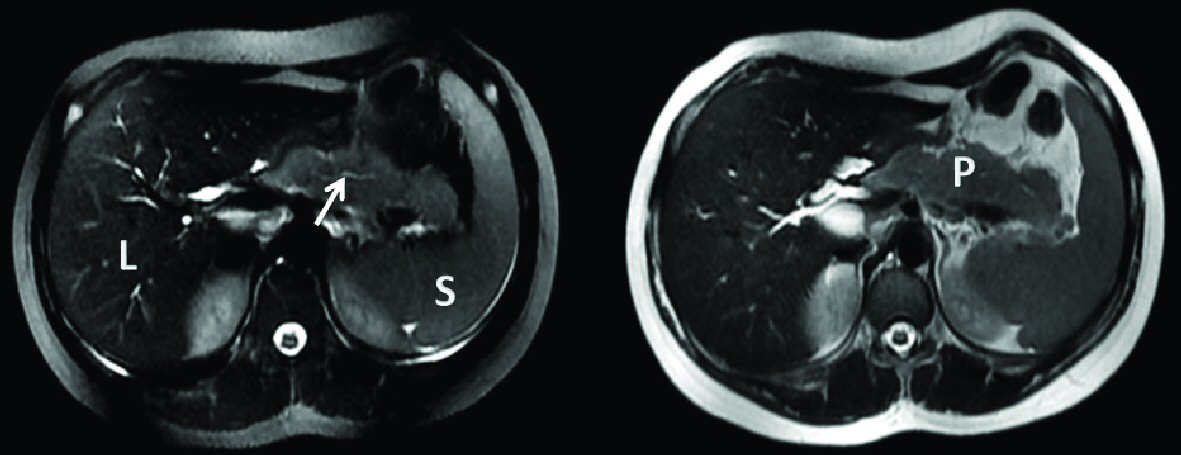 Click for large image | Figure 1. Axial T2 magnetic resonance cholangiopancreatography showing hypertrophied pancreas and irregular main pancreatic duct (arrow: narrow and irregular main pancreatic duct). L: liver; S: spleen; P: hypertrophied pancreas. |
One year later he presented with inflammatory anemia and rectal bleeding. Colonoscopy showed endoscopic and pathologic aspects that were compatible with ulcerative colitis (Fig. 2). He was treated first with mesalazine then with azathioprine and prednisone with improvement of symptoms. Prednisone was discontinued after a total of a 3-month treatment. Azathioprine was continued and the patient remained in remission for both pancreatitis and ulcerative colitis.
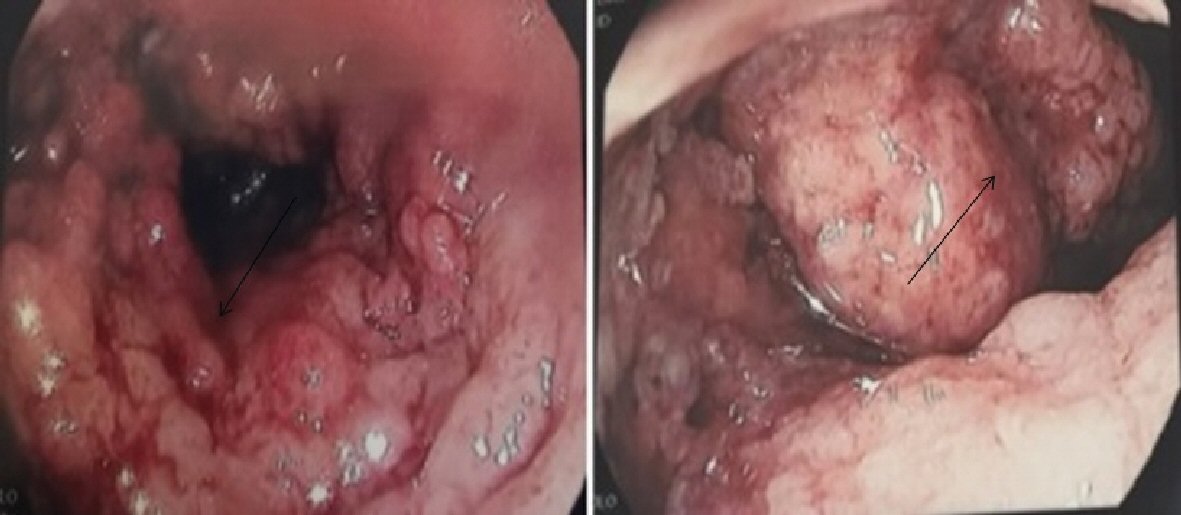 Click for large image | Figure 2. Macroscopic inflammatory aspect of the bowel compatible with ulcerative colitis (arrows). |
Case 2
A 12-year-old Lebanese Armenian boy, known to have ascendant aortic ectasia with no other health issues, was diagnosed with acute pancreatitis 6 days after aortic valve replacement. Physical exam showed heart murmur with normal abdominal and neurological exam. Medications received following the surgery included furosemide 2 mg/kg/day every 6 h, captopril 1 mg/kg/day every 12 h, vancomycin 60 mg/kg/day every 6 h, heparin 500 U/kg/day in continuous rate infusion, acenocoumarol 1 mg once daily and cefazolin 75 mg/kg/day every 8 h. Workup showed a lipase level at 1,297 IU/L, amylase at 294 IU/L, hemoglobin at 10.4 g/dL, WBC at 8,400/mm3, platelets at 458,000/mm3, ASAT at 26 IU, ALAT at 22 IU and creatinine at 40 µmol/L. He was kept NPO (except for oral medication) and received parenteral nutrition. Multiple viral serologies were negative. Calcium and triglyceride levels were normal. While the patient was under parenteral nutrition, he continued to present persistent abdominal pain and vomiting. Oral treatments were discontinued. Abdominal computed tomography (CT) scan showed a mild dilation of the gallbladder with normal pancreas and absence of dilation of the hepatic ducts. Parenteral nutrition was discontinued, liquid diet was started and acenocoumarol treatment was resumed. Two days later, the child presented fever and abdominal pain, with blood tests showing a rapid increase of lipase to a level of 2,561 IU/L. Hypothesis of drug-related pancreatitis secondary to acenocoumarol was held; hence, acenocoumarol was stopped. Lipase and amylase levels decreased and he was discharged with amylase at 247 IU/L and lipase at 1,201 IU/L.
One week after discharge from the hospital, despite the absence of abdominal pain or vomiting, an elevation of lipase to 2,400 IU/L was detected. He was readmitted to the hospital. Blood workup for autoimmune diseases showed no particularities with absence of autoantibodies (antinuclear antibodies, P-ANCA, C-ANCA, mitochondrial antibodies, anti-LKM1, anti-smooth muscle antibodies). The levels of complement (C3 and C4) were normal. Level of IgG4 was also within normal ranges at 99 mg/dL. Endoscopic ultrasound showed biliary sludge with no dilation of hepatic ducts. There was a hypertrophy and sausage shape of the pancreas suggestive of an AIP. The patient was treated with prednisone with a 1 mg/kg/day dose. He showed a favorable response to treatment and decrease in amylase and lipase levels. One month later, lipase and amylase dropped to normal levels. Prednisone dosage was decreased progressively with a total treatment of 4 months. The patient did not show any relapse. He did not show any clinical signs of pancreatitis, with periodic blood tests showing pancreatic enzymes within normal ranges for the 6 months to follow (with lipase reaching a level of 82 IU/L and amylase 61 IU/L 6 months after treatment).
| Discussion | ▴Top |
Although AIP is rarely seen in children as compared to adults, and is usually underdiagnosed, the introduction of the ICDC helped raising the index of suspicion for AIP thus allowing earlier diagnosis possibly without the full spectrum of pancreatic manifestations. Patients with AIP can present with variable symptoms including recurrent episodes of epigastric pain with rarely signs of acute pancreatitis such as nausea, vomiting and weight loss, or symptoms of other associated diseases [3, 7, 8]. Jaundice, acholia and choluria may be present in up to 80% of patients [7, 9]. Although jaundice is present in a high percentage of patients, our two patients did not present any jaundice, but only abdominal pain, nausea, or vomiting.
Many biological disorders may be found in association with AIP. Increased levels of pancreatic enzymes, hepatobiliary enzymes, bilirubin, and IgG are the main findings [1, 8, 10]. Various autoantibodies can be found but their presence is not specific for the disease [1, 8]. Autoantibodies may be found in 28 to 100% of cases, and their frequencies differ according to the type of antibody and the population studied [11]. According to one study, anti-nuclear antibodies and rheumatoid factor were positive in 44% and 16% of cases respectively [12]. One of the criteria for the diagnosis of AIP is elevated serum IgG4 level (> 140 mg/dL). In one study, IgG4 were found in 80% of patients with AIP type 1 and in 17% of patients with AIP type 2 [13]. However serum IgG4 can be elevated in multiple other conditions including cholangitis and pancreatic cancer [14]. Both patients had only isolated elevation of pancreatic enzymes levels with all other biological features within normal ranges. No autoantibodies were found and IgG4 were normal in both cases.
AIP can be associated with many other autoimmune diseases. Ulcerative colitis is more commonly associated with type 2 AIP, but can also be found in type 1 [5]. Furthermore, inflammatory bowel disease can be found in up to 14% of patients diagnosed with AIP [1, 8, 15]. The most frequent extrapancreatic lesion associated with AIP is sclerosing cholangitis.
Imaging techniques to study the morphology of the pancreas, the pancreatic and biliary ducts, constitute the cornerstone for diagnosis of AIP. Ultrasound usually shows dilated and sometimes thickened intrahepatic and extrahepatic biliary ducts, with enlarged and heterogeneous pancreatic parenchyma [9, 16]. Endoscopic retrograde pancreatography or MRCP can also show the enlarged pancreas with a better illustration of the narrowing of the main pancreatic duct. Diffuse enlargement of the pancreas with multiple stenosis of the main pancreatic duct without pancreatic stones is a pathognomonic radiological sign of AIP [1, 10]. In our both cases, imaging studies showed enlarged and heterogeneous pancreatic parenchyma with an irregular and narrow aspect of the main pancreatic duct. These signs were highly suggestive of AIP in our two patients.
Although seldom required, the diagnosis of AIP can be confirmed by obtaining histological evidence of an autoimmune process [17]. In type 1 AIP, these findings include lymphoplasmacytic infiltrate with storiform fibrosis and obliterative phlebitis (LPSP) or lymphoplasmacytic infiltrate with storiform fibrosis showing abundant (≥ 10 cells/HPF) IgG4-positive cells [18]. In type 2 AIP, granulocytic epithelial lesions can be found [19]. Pancreatic histology obtained by biopsy can be considered as gold standard for the diagnosis of AIP especially for the differential diagnosis with pancreatic cancer [20].
Steroids help improve clinical symptoms in first place, then biological disorders in about 2 weeks and finally morphological anomalies of the pancreas. Response to steroids treatment is included in the ICDC and is considered an important criterion when it shows improvement or resolution of radiological features obtained at baseline [6, 20]. The recommended starting dose is 1 mg/kg/day followed by progressive tapering of the dose [1]. Our first patient had a good control for ulcerative colitis with no recurrence of AIP features after steroid treatment. Our second patient presented resolution of signs and symptoms of AIP after receiving steroids.
In summary, the diagnosis of AIP for our first patient was suspected after the imaging results, then the presentation of ulcerative colitis and response for steroid therapy were in favor of the diagnosis. As for the second patient, the diagnosis of AIP was made upon the imaging results and response to steroids.
Conclusions
In conclusion, AIP is a rare disease in children, and a high index of suspicion must be present to diagnose it and differentiate it from other conditions. Established criteria help confirming the diagnosis, keeping in mind that IgG4 antibodies are associated with type 1 and autoantibodies are not specific. The pancreas is usually enlarged on imaging studies and the main pancreatic duct is narrow. Steroid therapy is the main treatment and response to steroids is included in the diagnostic criteria. It is important to highlight that an accurate diagnosis should be made in order to treat the condition and to avoid unnecessary surgical procedures when AIP is mistaken for pancreatic cancer, especially in adult patients, where both conditions can present as a pancreatic mass.
Grant Support
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Conflict of Interest
Authors have no conflict of interest to declare.
| References | ▴Top |
- Gargouri L, Ponsot P, Viala J, Belarbi N, Martinez C, Bellaiche M, Mougenot JF, et al. Recurrent autoimmune pancreatitis in a 10-year-old boy. J Pediatr Gastroenterol Nutr. 2009;48(3):374-377.
doi pubmed - Fukumori K, Shakado S, Miyahara T, Fukuizumi K, Takemoto R, Nishi H, Sakai H, et al. Atypical manifestations of pancreatitis with autoimmune phenomenon in an adolescent female. Intern Med. 2005;44(8):886-891.
doi pubmed - Ectors N, Maillet B, Aerts R, Geboes K, Donner A, Borchard F, Lankisch P, et al. Non-alcoholic duct destructive chronic pancreatitis. Gut. 1997;41(2):263-268.
doi pubmed - Cazares Camacho AG, Maldonado M, Faugier E, de Lourdes Cabrera M. Case autoimmune pancreatitis in children. Pediatr Rheumatol Online J. 2014;12(Suppl 1):P287.
doi - Bolia R, Chong SY, Coleman L, MacGregor D, Hardikar W, Oliver MR. Autoimmune Pancreatitis and IgG4 Related Disease in Three Children. ACG Case Rep J. 2016;3(4):e115.
doi - Shimosegawa T, Chari ST, Frulloni L, Kamisawa T, Kawa S, Mino-Kenudson M, Kim MH, et al. International consensus diagnostic criteria for autoimmune pancreatitis: guidelines of the International Association of Pancreatology. Pancreas. 2011;40(3):352-358.
doi pubmed - Okazaki K, Uchida K, Chiba T. Recent concept of autoimmune-related pancreatitis. J Gastroenterol. 2001;36(5):293-302.
doi pubmed - Nahon Uzan K, Levy P, O'Toole D, Belmatoug N, Vullierme MP, Couvelard A, Ponsot P, et al. Is idiopathic chronic pancreatitis an autoimmune disease? Clin Gastroenterol Hepatol. 2005;3(9):903-909.
doi - Blejter J, Weller S, Pace R, Cusumano H, Giambini D. Autoimmune pancreatitis: an adolescent case and review of literature. J Pediatr Surg. 2008;43(7):1368-1372.
doi pubmed - Horiuchi A, Kawa S, Akamatsu T, Aoki Y, Mukawa K, Furuya N, Ochi Y, et al. Characteristic pancreatic duct appearance in autoimmune chronic pancreatitis: a case report and review of the Japanese literature. Am J Gastroenterol. 1998;93(2):260-263.
doi pubmed - Smyk DS, Rigopoulou EI, Koutsoumpas AL, Kriese S, Burroughs AK, Bogdanos DP. Autoantibodies in autoimmune pancreatitis. Int J Rheumatol. 2012;2012:940831.
doi pubmed - Yamamoto T, Nakamura Y, Nishide J, Emi M, Ogawa M, Mori T, Matsubara K. Molecular cloning and nucleotide sequence of human pancreatic secretory trypsin inhibitor (PSTI) cDNA. Biochem Biophys Res Commun. 1985;132(2):605-612.
doi - Zhang L, Smyrk TC. Autoimmune pancreatitis and IgG4-related systemic diseases. Int J Clin Exp Pathol. 2010;3(5):491-504.
pubmed - Galloway DP, Wallihan D, Smith MT, Abu-El-Haija M. An unusual presentation of pediatric autoimmune pancreatitis. Pancreas. 2016;45(2):e1-2.
doi pubmed - Ohara H, Nakazawa T, Sano H, Ando T, Okamoto T, Takada H, Hayashi K, et al. Systemic extrapancreatic lesions associated with autoimmune pancreatitis. Pancreas. 2005;31(3):232-237.
doi pubmed - Takahashi N, Kawashima A, Fletcher JG, Chari ST. Renal involvement in patients with autoimmune pancreatitis: CT and MR imaging findings. Radiology. 2007;242(3):791-801.
doi pubmed - Refaat R, Harth M, Proschek P, Lindemayr S, Vogl TJ. Autoimmune pancreatitis in an 11-year-old boy. Pediatr Radiol. 2009;39(4):389-392.
doi pubmed - Autoimmune pancreatitis: an update [Internet]. Medscape. [cited 2017 Jul 1]. Available from: http://www.medscape.com/viewarticle/708921.
- Maruyama M, Watanabe T, Kanai K, Oguchi T, Muraki T, Hamano H, Arakura N, et al. International consensus diagnostic criteria for autoimmune pancreatitis and its Japanese amendment have improved diagnostic ability over existing criteria. Gastroenterol Res Pract. 2013;2013 :456965.
doi pubmed - Noarniakowska M, Kołodziejczyk E, Piwczynska K, Oracz G. Autoimmune pancreatitis in a 13.5-year-old child - a case report. Postępy Nauk Med. 2016;4:238-240.
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
International Journal of Clinical Pediatrics is published by Elmer Press Inc.