
| International Journal of Clinical Pediatrics, ISSN 1927-1255 print, 1927-1263 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, Int J Clin Pediatr and Elmer Press Inc |
| Journal website http://www.theijcp.org |
Case Report
Volume 6, Number 3-4, December 2017, pages 42-45
Microcephaly-Lymphedema-Chorioretinal Dysplasia Syndrome: Two Case Reports
Arzu Ekicia, f, Kadri Karaerb, Sevgi Yimeniciogluc, Kursat Bora Carmand, Mehmet Ali Ekicie
aDepartment of Pediatric Neurology, Bursa Yuksek Ihtisas Training and Research Hospital, Bursa, Turkey
bDepartment of Genetics, Dr. Ersin Arslan Training and Research Hospital, Gaziantep, Turkey
cDepartment of Pediatric Neurology, Eskisehir State Hospital, Eskisehir, Turkey
dDepartment of Pediatric Neurology, Osmangazi University, Medical School, Eskisehir, Turkey
eDepartment of Neurosurgery, Muradiye State Hospital, Bursa, Turkey
fCorresponding Author: Arzu Ekici, Department of Pediatric Neurology, Bursa Yuksek Ihtisas Training and Research Hospital, Mimar Sinan Ward., Emniyet Avenue, 16300 Bursa, Turkey
Manuscript submitted April 15, 2017, accepted July 26, 2017
Short title: MLCRD Syndrome
doi: https://doi.org/10.14740/ijcp275w
| Abstract | ▴Top |
Microcephaly-lymphedema-chorioretinal dysplasia is a rare syndrome in which the component of chorioretinopathy may develop later. We describe two patients with microcephaly-lymphedema-chorioretinal dysplasia syndrome who had characteristic facial features and congenital heart defects. Second patient had also lissencephaly which was very rarely reported before.
Keywords: Chorioretinopathy; Congenital heart defects; Lymphedema; Microcephaly; Lissencephaly
| Introduction | ▴Top |
Microcephaly-lymphedema-chorioretinal dysplasia (MLCRD) (OMIM #152950) is a rare syndrome. Previously it was described as microcephaly-lymphedema and microcephaly-chorioretinal dysplasia. It has been suggested that both of them are the same entity [1]. The mode of inheritance is uncertain. Both sporadic and familial cases were described [2]. Herein we describe two patients with MLCRD who had characteristic facial features and congenital heart anomalies. Second patient had also lissencephaly which was very rarely reported before.
| Case Reports | ▴Top |
Case 1
A 3-month-old girl was admitted to our hospital with the complaint of microcephaly. She was
the second child and her parents were non-consanguineous. She was born following a spontaneous vaginal delivery at term. The infant’s weight was 3,370 g (50th-75th percentile), and head circumference was 31.5 cm (< 5th percentile) at birth. Apgar scores were 9 (1 min) and 10 (5 min).
On physical examination, body weight was 4,390 g (10th-25th percentile), length was 59 cm (10-25 percentile), and head circumference was 34.6 cm (< 5th percentile). She was severely microcephalic and anterior fontanel was closed. She had a facial appearance with broad nose with rounded tip, long philtrum with thin upper lip and prominent ears broad (Fig. 1a). She had a grade 2/6 systolic heart murmur and bilateral edema of the dorsum of the feet (Fig. 1b). Neurologic examination and initial ophthalmic examination were normal.
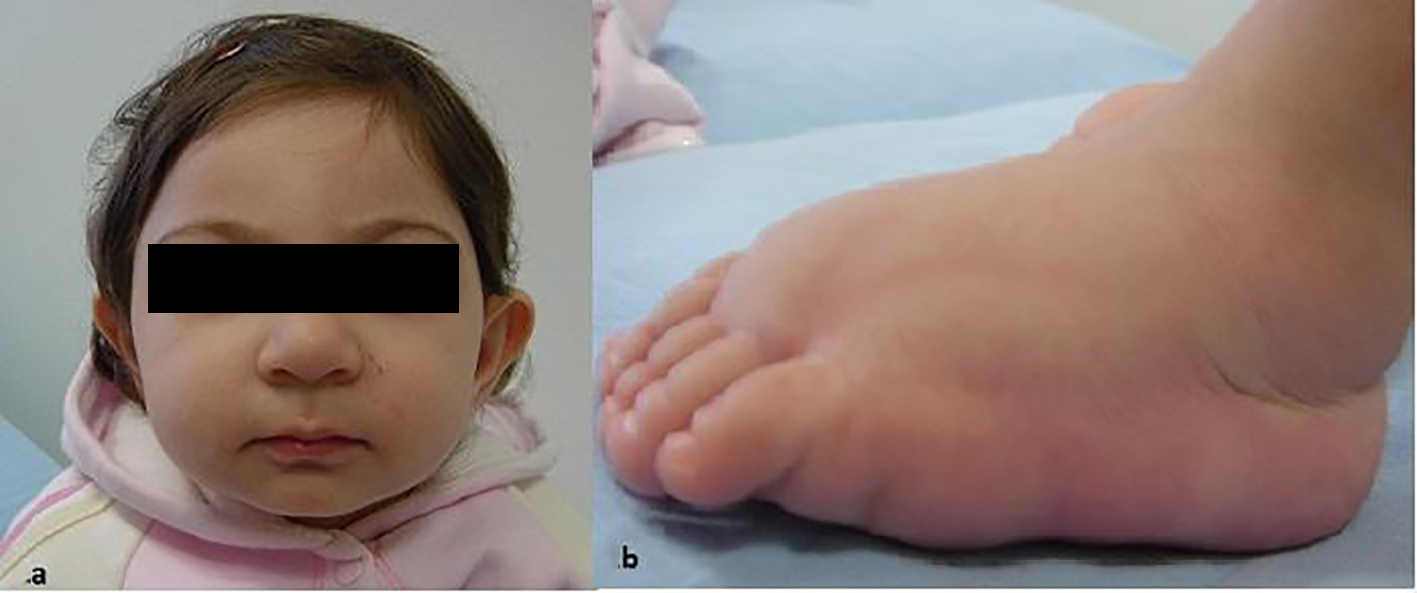 Click for large image | Figure 1. Case 1: (a) characteristic facial features; (b) lymphedema. |
Her developmental milestones were appropriate of her age. Her ophthalmic examination at 9 months of age showed bilateral chorioretinal changes and retinal pigmentation. She had no visual deterioration. Visual-evoked potentials showed optic nerve dysfunction.
Laboratory investigations revealed that serum electrolytes, serum protein and albumin levels total blood count, thyroid hormones, serology for congenital infections, serum and urinary amino acids, serum lactate, pyruvate and ammonium were normal. Cytogenetic analysis of her peripheral blood indicated a normal 46,XX karyotype. Cardiac echocardiography (ECO) revealed a secundum atrial septal defect. Cranial magnetic resonance imaging (MRI) was normal except microcephaly.
Her physical examination at 17 months of age showed head circumference of 40 cm (< 5th percentile). She still has bilateral edema of the feet which diminished. She is saying about 20 words. Her mental motor development is normal at 2 years of age.
Case 2
A 4-month-old boy was admitted to our hospital with the complaint of microcephaly. He was the second child of healthy non-consanguineous parents. His mother had uterine bleeding at 24 weeks’ gestation. The ultrasound examination showed intrauterine growth retardation and placental blood flow was reduced. The mother had used acetylsalicylic acid and subcutaneous low molecular weight heparin after 24 weeks of gestation.
He was born at 39 weeks of gestation by cesarean section. Apgar scores were 9 (1 min) and 10 (5 min). Birth weight was 2,500 g (2th-5th percentile), length was 52 cm (90th-95th percentile), and head circumference was 30.5 cm (< 5th percentile). Both parents have a normal head size and no family history of microcephaly or mental retardation.
On examination at 4 months of age, body weight was 5,100 g (10-25th percentile), length was 63 cm (75th percentile), and head circumference was 35 cm (< 5th percentile). He had broad nose with rounded tip and long philtrum with thin upper lip. The anterior fontanel size was 1 × 1 cm. A pansystolic murmur of grade 2/6 was detected. He had bilateral edema of the feet. He was able to hold his head. The fundoscopic examination showed optic atrophy and central retinal vessels atrophy. There was diffuse patchy chorioretinal atrophy at the peripheric retina and choroid vessels can be seen at these areas (Fig. 2). He had nystagmus and poor eye contact. He did not come for his follow-up. Laboratory investigations revealed that serum electrolytes, serum protein, serum albumin levels, total blood count, thyroid hormones, TORCH screening, serum and urinary amino acids, serum lactate, pyruvate and ammonium were normal. Abdominal ultrasonography was normal. The echocardiogram showed ventricular septal defect. Cranial MRI showed lissencephaly (Fig. 3).
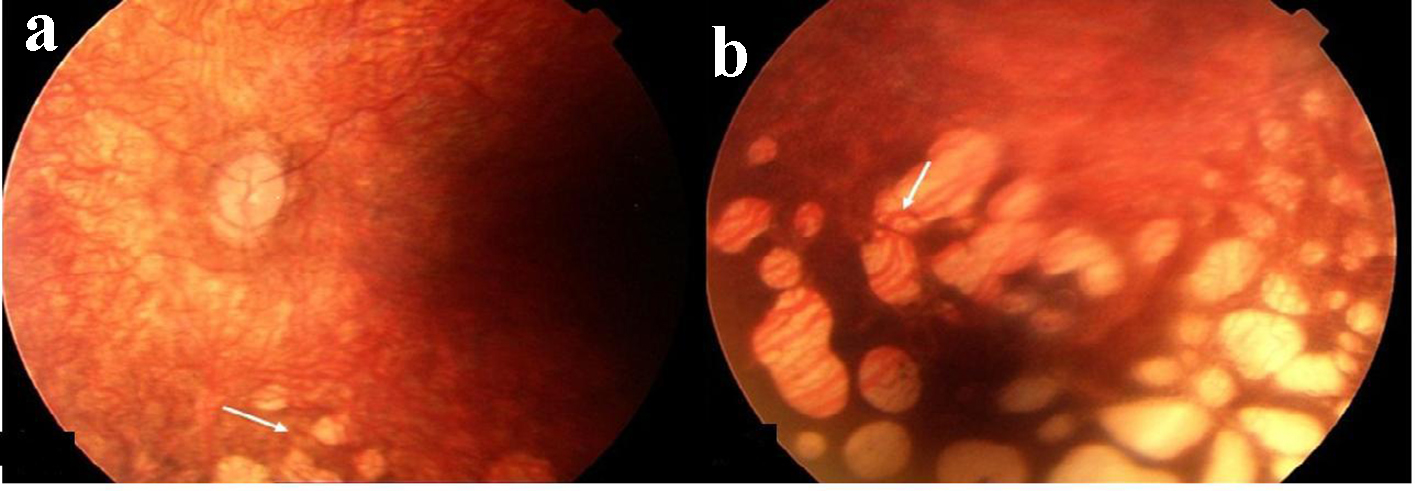 Click for large image | Figure 2. Case 2: (a) optic atrophy and central retinal vessels atrophy; (b) diffuse patchy peripheric chorioretinal atrophy and choroid vessels. |
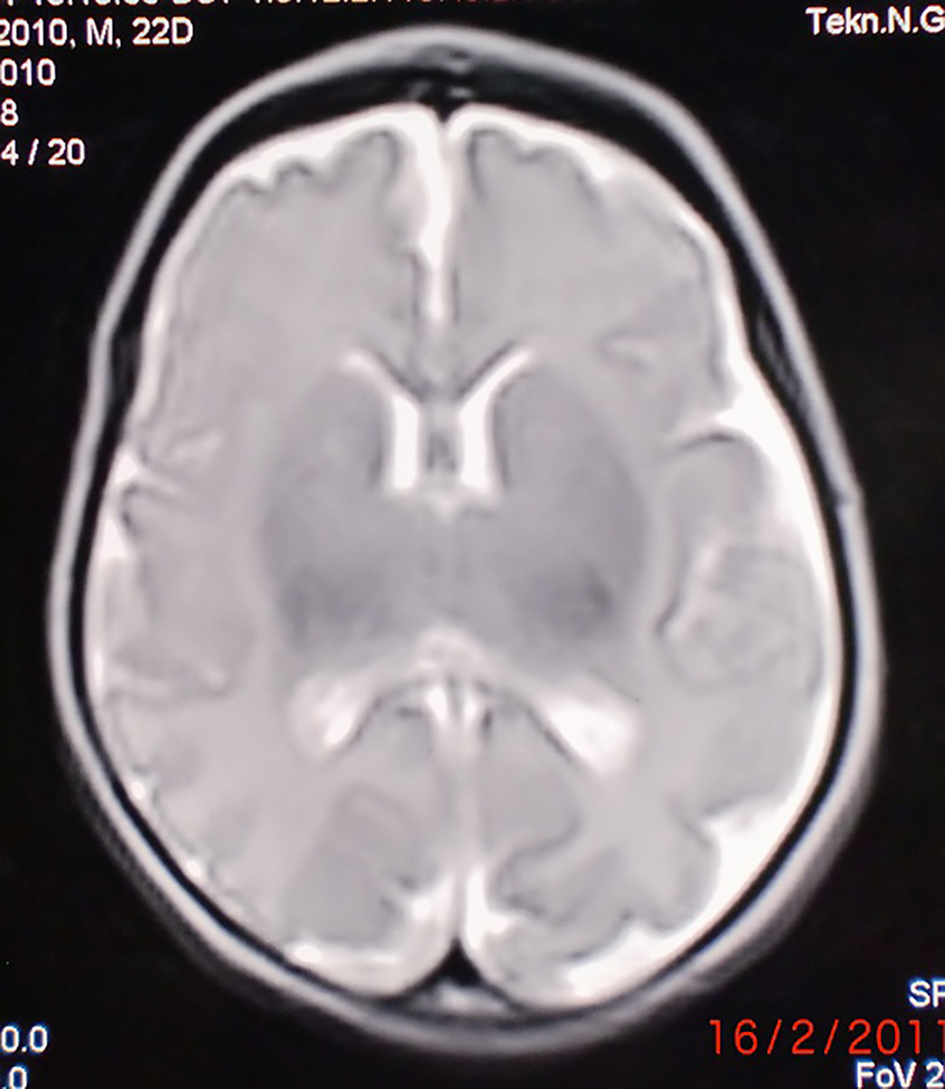 Click for large image | Figure 3. Case 2: MRG showed microcephaly and microlissencephaly. |
| Discussion | ▴Top |
Microcephaly might be seen with several syndromes. The patient with microcephaly must be searched for the other system anomalies such as lymphedema and ocular findings. Congenital lymphedema was well-known features of Turner syndrome, Noonan syndrome and the autosomal dominantly inherited Milroy disease [3, 4]. Our two patients had bilateral edema of the feet. Lymphedema is a component of MLCRD syndrome which is rarely seen, can be seen at birth in only feet or all four limbs and can diminish by time [3, 4].
The description of a syndrome encompassing microcephaly and chorioretinal dysplasia was first mentioned by Mc Kusick [5]. Additionally, microcephaly and lymphedema (MIM152950) was first described by Leung in 1985 in five individuals in a four generation family and ascribed it to be a dominantly inherited syndrome [6]. There was no mental retardation. Furthermore, the high degree of intrafamilial variability of these syndromes reported underscores the need to fully evaluate parents and siblings of all affected individuals [7]. Based on the variability of features observed in some families, several authors have argued that autosomal dominant microcephaly with chorioretinopathy and the lymphedema, microcephaly, and chorioretinopathy syndrome may represent variable expressions of the same entity. Ostergaard et al [8] analyzed the KIF11 gene in unrelated chorioretinal dysplasia, microcephaly and mental retardation syndrome (CDMMR) and MLCRD syndrome families identified heterozygous mutations in KIF11 gene. It was concluded that the MLCRD and CDMMR syndromes should be considered a single entity with variable clinical features. They can be observed as an autosomal dominant disorder with variable expressivity, mainly characterized by mild to severe microcephaly, often associated with developmental delay, ocular defects and lymphedema, essentially on the dorsum of the feet [10].
Ophthalmological findings reported in the autosomal dominant syndrome of MLCRD include chorioretinal dysplasia, myopic astigmatism, and retinal dystrophy. Chorioretinal dysplasia is the most common ophthalmic abnormality. And also peripheral retinal pigmentation, retinal folds, optic atrophy, and macular damages can be seen. The ocular involvement may develop later; therefore, the ophthalmologic follow-up is required. In our first case chorioretinal dysplasia was detected at the second fundoscopic examination. If there were retinal folds, optic atrophy or macular involvement, severe myopia, nystagmus or blindness can occur. Otherwise the patients with chorioretinal dysplasia can have stable vision [9]. In our first case, she had chorioretinal lesions and retinal pigmentation and her visual performance was good. Unfortunately our second patient had poor eye contact and nystagmus that had diffuse chorioretinal and optic atrophy.
Our two patients had similar facial features. Vasudevan et al [1] suggested that typical facial features of MLCRD syndrome was upslanting palpebral fissures, broad nose with rounded tip, anteverted nares, long philtrum with thin upper lip, pointed chin, and prominent ears. Congenital heart defects can be frequently seen in MLCRD [3, 9, 10]. Both of our patients have congenital heart defect such as ASD and VSD. Congenital heart defects might be more frequent than reported before. Therefore echocardiography must be done at all MLCRD patients. Cortical malformations, also cortical dysplasia might be a part of the spectrum of this syndrome. Brain imaging demonstrated that simple microcephaly in patients with MLCRD syndrome [10]. In our second case we demonstrate lissencephaly which is rarely reported.
No universal criteria exist to delineate the microcephaly and chorioretinopathy disorders. Classification has been variably based on the presence or absence of lymphedema, the existence or lack of mental retardation, the type of chorioretinal lesion or dystrophy, the constellation of dysmorphic features, and the apparent inheritance pattern observed in the family. Counselling in microcephaly is difficult, and in the absence of a specific etiological diagnosis, an empirical recurrence risk of 15-20% is often cited [11].
In summary, patients with microcephaly should be evaluated in terms of other organ involvement. Here we describe two MLCRD patients with congenital heart defects and lissencephaly in case 2. In order to demonstrate chorioretinopathy, fundoscopic examination should be performed in patients with microcephaly and lymphedema. And also it must be known that the ocular involvement may develop later.
Conflict of Interest
The authors disclosed no conflict of interest during the preparation or publication of this manuscript.
Grant Support
The authors disclosed that they did not receive any grant during conduction or writing of this study.
| References | ▴Top |
- Vasudevan PC, Garcia-Minaur S, Botella MP, Perez-Aytes A, Shannon NL, Quarrell OW. Microcephaly-lymphoedema-chorioretinal dysplasia: three cases to delineate the facial phenotype and review of the literature. Clin Dysmorphol. 2005;14(3):109-116.
doi pubmed - Eventov-Friedman S, Singer A, Shinwell ES. Microcephaly, lymphedema, chorioretinopathy and atrial septal defect: a case report and review of the literature. Acta Paediatr. 2009;98(4):758-759.
doi pubmed - Strauss RM, Ferguson AD, Rittey CD, Cork MJ. Microcephaly-lymphoedema-chorioretinal-dysplasia syndrome with atrial septal defect. Pediatr Dermatol. 2005;22(4):373-374.
doi pubmed - Trzupek KM, Falk RE, Demer JL, Weleber RG. Microcephaly with chorioretinopathy in a brother-sister pair: evidence for germ line mosaicism and further delineation of the ocular phenotype. Am J Med Genet A. 2007;143A(11):1218-1222.
doi pubmed - McKusick VA, Stauffer M, Knox DL, Clark DB. Chorioretinopathy with hereditary microcephaly. Arch Ophthalmol. 1966;75(5):597-600.
doi pubmed - Leung AK. Dominantly inherited syndrome of microcephaly and congenital lymphedema. Clin Genet. 1985;27(6):611-612.
doi pubmed - Sadler LS, Robinson LK. Chorioretinal dysplasia-microcephaly-mental retardation syndrome: report of an American family. Am J Med Genet. 1993;47(1):65-68.
doi pubmed - Ostergaard P, Simpson MA, Mendola A, Vasudevan P, Connell FC, van Impel A, Moore AT, et al. Mutations in KIF11 cause autosomal-dominant microcephaly variably associated with congenital lymphedema and chorioretinopathy. Am J Hum Genet. 2012;90(2):356-362.
doi pubmed - Limwongse C, Wyszynski RE, Dickerman LH, Robin NH. Microcephaly-lymphedema-chorioretinal dysplasia: a unique genetic syndrome with variable expression and possible characteristic facial appearance. Am J Med Genet. 1999;86(3):215-218.
doi - Casteels I, Devriendt K, Van Cleynenbreugel H, Demaerel P, De Tavernier F, Fryns JP. Autosomal dominant microcephaly--lymphoedema-chorioretinal dysplasia syndrome. Br J Ophthalmol. 2001;85(4):499-500.
doi pubmed - Mahmoud AA, Abdul-Aziz MM, Al-Tala SM, Ahmed FO. Microcephaly, retinal dysplasia, pedal edema, mental retardation, and short stature. Neurosciences (Riyadh). 2006;11(3):210-212.
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
International Journal of Clinical Pediatrics is published by Elmer Press Inc.