International Journal of Clinical Pediatrics, ISSN 1927-1255 print, 1927-1263 online, Open Access
Article copyright, the authors; Journal compilation copyright, Int J Clin Pediatr and Elmer Press Inc
Journal website http://www.theijcp.org
Case Report
Volume 6, Number 1-2, June 2017, pages 28-32
A Patient With Focal Dermal Hypoplasia Syndrome and Renal Involvement due to a Novel Mutation in the PORCN Gene
Luis E. Becerra-Solanoa, c, Liliana Chaconb, Dinorah Morales-Matab, Efrain Monroy-Marquezb, Eunice Lopez-Munozc, Jose E. Garcia-Ortizd, e, f
aUnidad de Investigacion Medica, Hospital de Pediatria, Instituto Mexicano del Seguro Social, Guadalajara, Jalisco, Mexico bCentro de Rehabilitacion Infantil Teleton, Chihuahua, Chihuahua, Mexico cUnidad de Investigacion Medica en Medicina Reproductiva, Hospital de Gineco-Obstetricia No. 4 “Luis Castelazo Ayala”, Instituto Mexicano del Seguro Social, Ciudad de Mexico, Mexico dDivision de Genetica, Centro de Investigacion Biomedica de Occidente, Instituto Mexicano del Seguro Social, Guadalajara, Mexico eDireccion de Educacion e Investigacion en Salud, Unidad Medica de Alta Especialidad Hospital de Gineco-Obstetricia, Instituto Mexicano del Seguro Social, Guadalajara, Mexico fCorresponding Author: Jose Elias Garcia-Ortiz, Centro de Investigacion Biomedica de Occidente, CMNO-IMSS, Sierra Mojada 800, Col. Independencia, CP 44340, Guadalajara, Jalisco, Mexico
Manuscript submitted May 25, 2017, accepted June 16, 2017 Short title: A PORCN Gene Mutation and Renal Involvement doi: https://doi.org/10.14740/ijcp277w
Focal dermal hypoplasia (FDH) is an uncommon X-linked dominant entity associated with mutations on the PORCN gene. FDH is characterized by cutaneous, skeletal, dental, ocular, and soft tissue defects. Here we report a female patient with an illustrative clinical case of FDH as well as renal malformations and a novel deleterious mutation on PORCN gene.
Goltz-Gorlin syndrome, also known as focal dermal hypoplasia (FDH), is a genodermatosis associated to mutations in PORCN gene (Xp11.23), with X-linked dominant inheritance [1, 2]. It is characterized by atrophic skin that follows the lines of Blaschko on both sides but not symmetrically, and asymmetrical involvement of hands and feet with split hand, syndactyly, polydactyly or clinodactyly [2, 3]. Ocular and dental anomalies have also been described [4].
Variability in clinical phenotype of FDH is attributed to degrees of mosaicism, more than random X-chromosomal inactivation [5, 6]. Occasionally, it shows without FDH [2].
In 95% of patients, the presentation is sporadic and most of them are females. Male patients are the result of postzygotic mosaicism or aneuploidies involving the X-chromosome [5].
Most mutations are occurring as somatic mosaicism after the postzygotic stage [1]. Due to mosaicism, diagnosis of FDH is difficult; in some cases, mutations are only found on fibroblast analysis through skin biopsy [3, 5].
Up to date, the web of LOVD for PORCN gene has recorded 119 unique DNA variants, 165 individuals with variant(s), and 171 variants [7].
The purpose of this article is to contribute to the knowledge of FDH describing a female patient with renal involvement (the reports of patient with Goltz syndrome and involvement of genitourinary system are scarce) and a novel mutation on the PORCN gene, who was included in study of 53 FDH patients by Fernandes et al [6]. Then, these findings permitted us to establish a genotype-phenotype correlation.
Up to date, there are only five reports of patients with Goltz syndrome and involvement of genitourinary system. Present case is important because a novel mutation is reported.
A female patient was evaluated at first time at 1 year 6 months of age by the medical genetics service due to microcephaly, microphthalmia, dermatoses, cardiac anomalies, and limb involvement. She was the product of healthy, non-consanguineous parents (mother 32 years, G-1, C-1; father 26 years). During the seventh month prenatal control, only placental calcification was detected by ultrasonography. The pregnancy continued without complications. The infant was born at 36 weeks of gestation by cesarean section due to premature rupture of membranes. Birth weight was 2,600 g, length was 47 cm, and Apgar score was 8 - 9. After birth, persistence of ductus arteriosus, vesicular urethral reflux, and iris coloboma on the left eye were diagnosed. She remained at the hospital for 10 days and received phototherapy for 2 days due to jaundice. Psychomotor development was normal. She received occupational and physical therapy to improve and develop her skills, then she is able to interact with other persons and to perform daily life activities by herself. Her psychological age is according to chronological age. At present she is egressed of CRIT-Chihuahua.
Physical examination
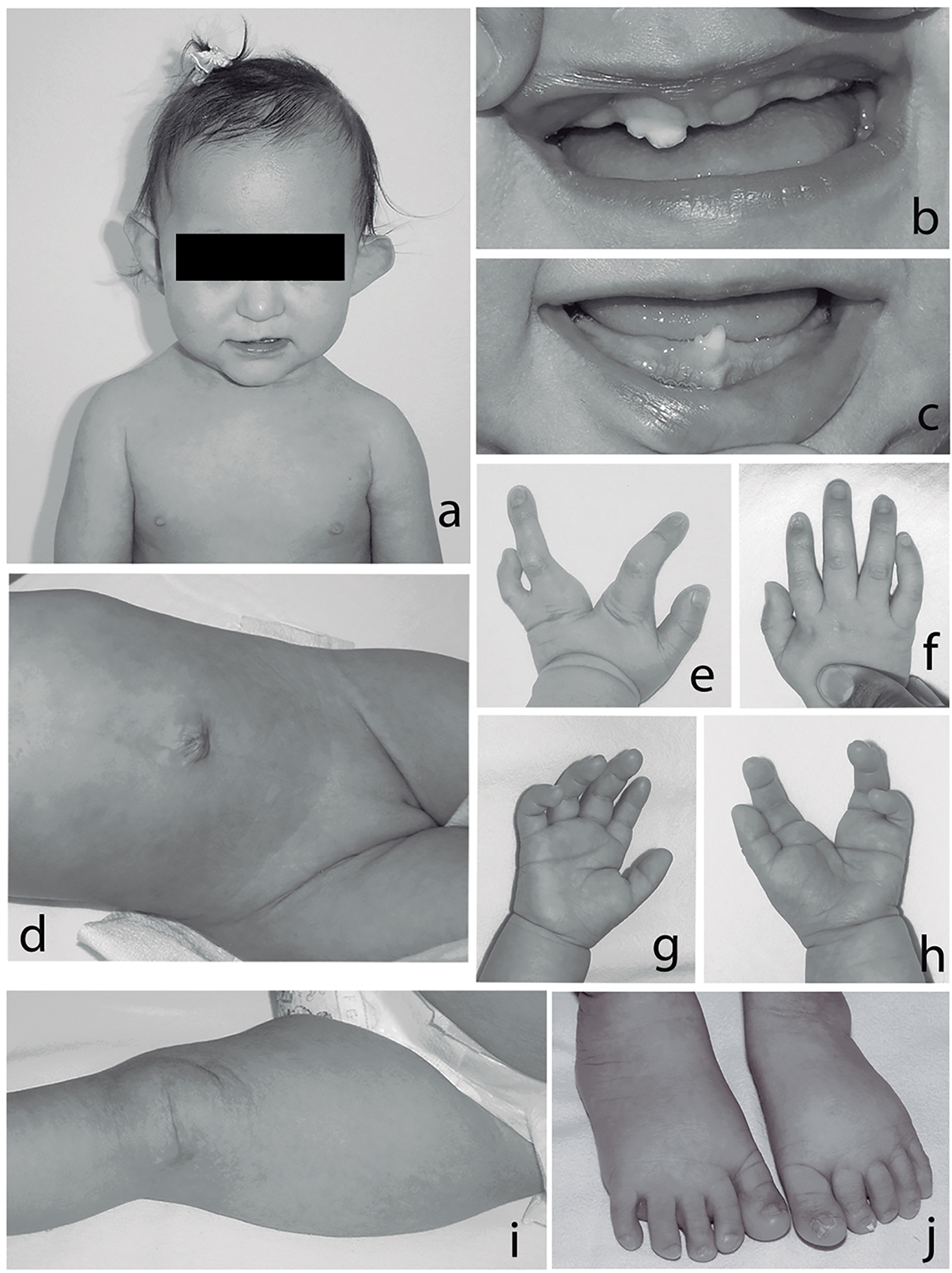
Anthropometry at 4 years of age demonstrated the following: weight 13 kg (-2.68 SD), height 103 cm (-0.82 SD), and occipitofrontal circumference 46 cm (-1 SD). Physical examination revealed microcephaly, sparse hair, hypotrichotic eyebrows, slanted palpebral fissures, microphthalmia and iris coloboma of the left eye. There was left facial hemiatrophy, high nasal bridge, downward nasal tip, thin upper lip with left asymmetry, oral cavity with dysplastic teeth and oligodontia, pointed chin, dysplastic anteverted and simplified helix, short neck, and wide-spaced nipples (Fig. 1a, b, c). Cardiac sounds were normal. Abdomen was without visceromegalies and with umbilical hernia. Genitalia demonstrated hypoplastic left labium majora (Fig. 1d). Limbs showed split hand on left hand, and right hand had modified flexion folds and dystrophic nails on right index finger (Fig. 1e, f, g, h). Left lower limb displayed asymmetry and dystrophic nails were shown on toes of both feet (Fig. 1j). Skin showed areas of subcutaneous fat herniation, atrophy and pigmented patchy aspect along the lines of Blaschko, more evident on the left side of the body (Fig. 1d, i).
Figure 1. (a) Sparse hair, facial asymmetry, hypotrichotic eye brushes, left microphthalmia, dysplastic and anteverted helix; (b, c) oligodontia, small teeth, and enamel abnormalities; (d) umbilical hernia, hypoplastic left labium majora; (e-h) split left hand, note brittle nail on second right finger, and modified palmar creases; (i) note patchy atrophic and pigmented skin; (j) hypoplastic and brittle nails on toes, second toes laterally deviated.
X-ray studies
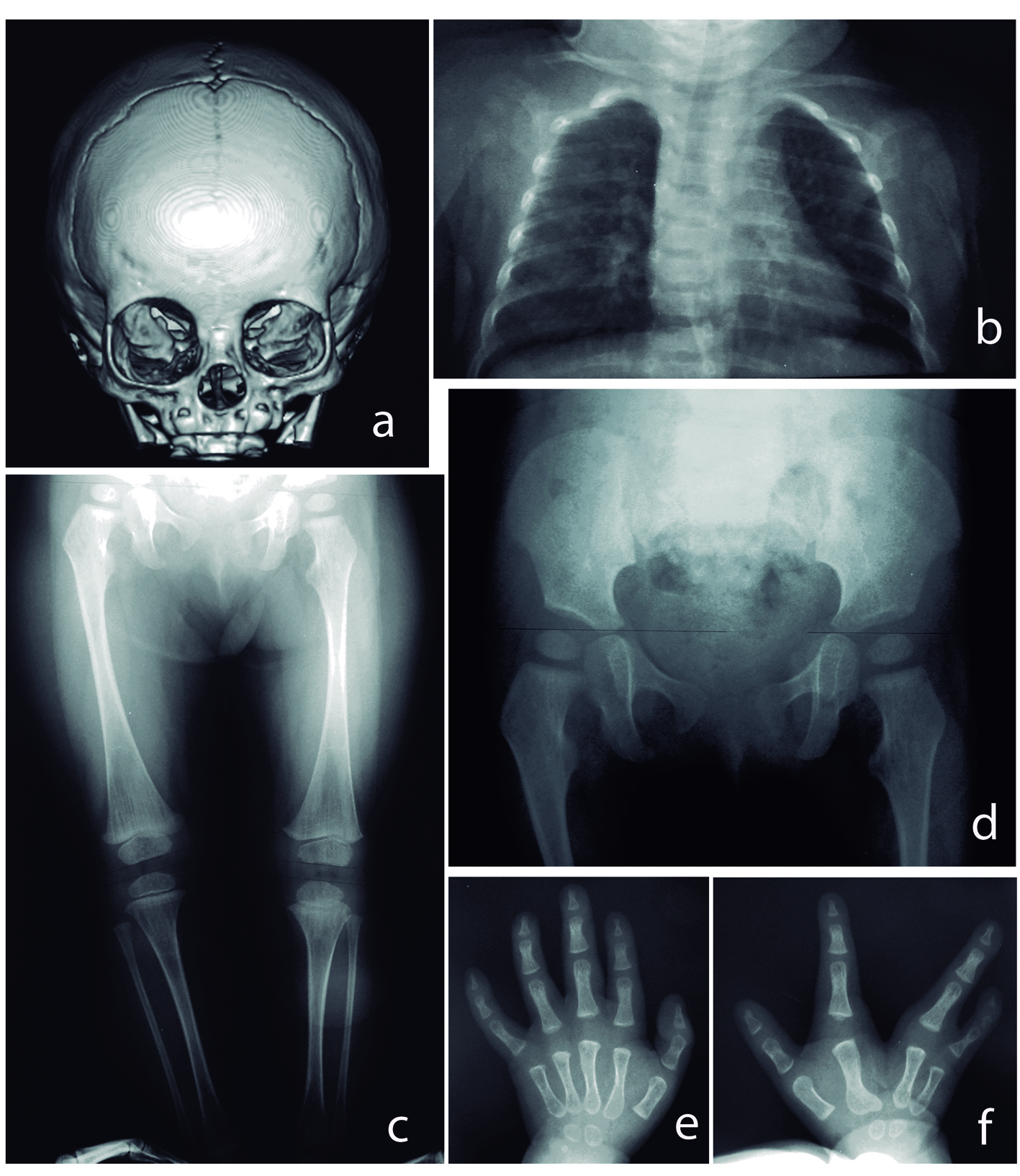
Tri-dimensional reconstruction of the skull disclosed asymmetric ocular orbits due to left hypoplasia (Fig. 2a). X-ray showed thorax with apparent asymmetry, slender long bones with thin cortical diaphysis and wide metaphysis (Fig. 2b, c). Femoral necks were rectified, and hypoplasia of distal epiphyseal nuclei and modified metaphysis was demonstrated in left tibia and fibula (Fig. 2d). Ossification of metacarpal bones was asymmetric (four nuclei on right side, and two nuclei on left side; 11 months old). Reduced bone density and osteopathia striata were also noticed (Fig. 2e, f). Bone scan of lower extremities revealed asymmetry of the left limb.
Figure 2. (a) Left skull asymmetry. (b) Thorax with no osseous abnormalities, apparently asymmetric thorax. (c) Asymmetry of lower extremities. Hypoplasia of distal epiphyseal nuclei on left limb was noted and modified metaphysis in left tibia and fibula, slender long bones, with thin cortical diaphysis, and wide metaphysis. Note osteopathia striata. (d) Rectified femoral necks. (e, f) Split right hand, asymmetric ossification of metacarpal bones (four nuclei on right side, and two nuclei on left side; 11 months old) and reduced bone density.
Computed tomography of the brain revealed only microcephaly. Magnetic resonance imaging of left ocular globe reported absence of scleral tissue due to coloboma. Right eye was structurally normal. Echocardiogram demonstrated persistence of foramen ovale with a left-right shunt, without hemodynamic repercussions. Pyelocaliceal ectasia in left kidney was also reported on abdominal ultrasonography.
Skin biopsy showed a cystic neoformation coated with stratified squamous epithelium with diffuse infiltration by lymphocytes and monocytes.
Laboratory tests
Karyotype (46,XX; GTG stained, at 450 - 550 bands), and screening test for metabolic disease were normal. Also, clinical chemical tests performed (glucose, urea, creatinine, uric acid, bilirubin, and urinalysis) were reported as normal.
The molecular analysis was performed in the Medical Genetics Laboratories, Baylor College, Houston, TX, USA. DNA was used for PCR amplification of exons 2-15 of the PORCN gene (GenBank accession number NM_203475.1, variant D). Primers for all exons flank the respective intron/exon junctions. Direct sequence analysis of PCR products was performed in both forward and reverse directions using automated fluorescence dideoxy sequencing methods. Base numbering refers to the A of the ATG start codon as position 1 [8, 9].
After molecular analysis, a novel mutation was found: c.1040T>C (p.Leu347Pro). Bioinformatic analysis was performed with PROVEAN protein (-6.69, cutoff -2.5; deleterious), SIFT prediction (0.001, cutoff 0.05; damaging), PolyPhen-2 (the predicted amino acid change has a score of 1, indicating that this is highly likely to be damaging to the structure and function of PORCN), and by tri-dimensional reconstruction using STRUM results in a ddG of 1.47 (tendency to destabilized protein). Additionally, the study was also performed on the mother and no mutations were found; DNA paternal was not performed; moreover, he had no clinical features related to FDH.
The patient reported here demonstrated clinical features involving in the most affected organs in FDH: dermatological, skeletal, cardiac, ophthalmic, and dental; additionally pyelocaliceal ectasia on the left kidney, and a novel mutation in the PORCN gene (c.1040T>C; p.Leu347Pro). This mutation confers a deleterious or damaging effect, and destabilized protein. Molecular modeling studies suggest that the L347P residue lies in the PORCN transmembrane domain, and this domain contributes to the functionality of PORCN [6]. In present patient, the molecular results and clinical features support a correlation genotype-phenotype.
Porcupine (Porcn) was discovered as Drosophila segment polarity gene necessary for the normal distribution of Wingless (Wg, Drosophila homolog of WNT). Studies performed in mouse model demonstrated that ectodermal-specific inactivation in the skin causes dermic and dental anomalies; also, inactivation in mesenchyme of limbs leads to shortening of long bones and digits. Moreover, cell-based assays demonstrated a reduction in secretion of WNT3A in the presence of mutant PORCN. The combined findings support that defective Wnt signaling is the basis of the phenotypic features of FDH. Also Porcupine regulates Wnt signaling during the development of the urogenital tract [1], and WNT4 has a critical role in the development of the reproductive system, kidneys, adrenal glands, pituitary gland, and mammary tissue [10]. In a study performed by Ross et al [11], they demonstrated that cells with defective PORCN gene were incapable to generate pluripotent stem cell from fibroblasts, concluding that deficient or inactivated PORCN cell cannot to activate WNT pathway. It is another evidence for relationship for interaction between PORCN and WNT. Then, association of WNT pathway to urogenital embryological development could explain malformation in this system.
To our knowledge, there is not previous report of pyelocaliceal ectasia in FDH. We only found five reports of genitourinary involvement: malrotation and possible horseshoe kidneys [4], one horseshoe kidney [12], one of renal hypoplasia [13], one of renal agenesis with uterus bicornis [14], and one of renal ectopia with uterus bicornis [15].
Differential diagnoses must include those syndromes that share dermal, skeletal, and ocular involvement, e.g., microphthalmia with linear skin defects (MLS) syndrome (OMIM 309801), Incontinentia pigmenti (OMIM 308310), Rothmund-Thomson syndrome (OMIM 268400), and MIDAS (MIDAS: microphthalmia, dermal aplasia, and sclerocornea syndrome; OMIM 309801).
In summary, this patient discloses typical clinical features of Goltz syndrome, moreover, an infrequent genitourinary system affectation. Until knowledge, even the participation of WNT pathway in embryological development of this system, there are few reports of genitourinary system involvement, so we think more patients with genitourinary system involvement exist.
Conclusion
There is an association between mutations in PORCN gene and FDH, and despite the absence of genotype-phenotype correlation in many patients, the clinical spectrum is almost constant. In present patient clinical manifestations and in silico analysis are indicative of genotype-phenotype correlation.
Considering the relationship between WNT pathway and urogenital developing, it might not be usual that malformations can be observed among FDH patients, until knowledge.
Disclosure
This article has no funding source, and the authors have no conflicts of interest to declare.
Liu W, Shaver TM, Balasa A, Ljungberg MC, Wang X, Wen S, Nguyen H, et al. Deletion of Porcn in Mice Leads to Multiple Developmental Defects and Models Human Focal Dermal Hypoplasia (Goltz Syndrome). PLoS One. 2012;7(3):e32331. doipubmed
Contreras-Capetillo SN, Lombardi MP, Pinto-Escalante D, Hennekam RC. Focal Dermal Hypoplasia without Focal Dermal Hypoplasia. Am J Med Genet A. 2014;164A(3):778-781. doipubmed
Sheu J, Divito SJ, Hoffman EP, Frank NY, Merola JF. Mosaic Focal Dermal Hypoplasia Caused by a Novel Somatic Mutation in Porcn Detected in Affected Skin. Br J Dermatol. 2015;173(2):568-570. doipubmed
Goltz RW, Henderson RR, Hitch JM, Ott JE. Focal Dermal Hypoplasia Syndrome. A Review of the Literature and Report of Two Cases. Arch Dermatol. 1970;101(1):1-11. doipubmed
Alkindi S, Battin M, Aftimos S, Purvis D. Focal Dermal Hypoplasia Due to a Novel Mutation in a Boy with Klinefelter Syndrome. Pediatr Dermatol. 2013;30(4):476-479. doipubmed
Fernandes PH, Wen S, Sutton VR, Ward PA, Van den Veyver IB, Fang P. Porcn Mutations and Variants Identified in Patients with Focal Dermal Hypoplasia through Diagnostic Gene Sequencing. Genet Test Mol Biomarkers. 2010;14(5):709-713. doipubmed
Fokkema IF, Taschner PE, Schaafsma GC, Celli J, Laros JF, den Dunnen JT. Lovd V.2.0: The Next Generation in Gene Variant Databases. Hum Mutat. 2011;32(5):557-563. doipubmed
Grzeschik KH, Bornholdt D, Oeffner F, Konig A, del Carmen Boente M, Enders H, Fritz B, et al. Deficiency of Porcn, a Regulator of Wnt Signaling, Is Associated with Focal Dermal Hypoplasia. Nat Genet. 2007;39(7):833-835. doipubmed
Wang X, Reid Sutton V, Omar Peraza-Llanes J, Yu Z, Rosetta R, Kou YC, Eble TN, et al. Mutations in X-Linked Porcn, a Putative Regulator of Wnt Signaling, Cause Focal Dermal Hypoplasia. Nat Genet. 2007;39(7):836-838. doipubmed
Ocal G. Current Concepts in Disorders of Sexual Development. J Clin Res Pediatr Endocrinol. 2011;3(3):105-114. doipubmed
Ross J, Busch J, Mintz E, Ng D, Stanley A, Brafman D, Sutton VR, et al. A Rare Human Syndrome Provides Genetic Evidence That Wnt Signaling Is Required for Reprogramming of Fibroblasts to Induced Pluripotent Stem Cells. Cell Rep. 2014;9(5):1770-1780. doipubmed
Suskan E, Kurkcuoglu N, Uluoglu O. Focal Dermal Hypoplasia (Goltz Syndrome) with Horseshoe Kidney Abnormality. Pediatr Dermatol. 1990;7(4):283-286. doipubmed
Landa N, Oleaga JM, Raton JA, Gardeazabal J, Diaz-Perez JL. Focal Dermal Hypoplasia (Goltz Syndrome): An Adult Case with Multisystemic Involvement. J Am Acad Dermatol. 1993;28(1):86-89. doi
Reddy J, Laufer MR. Congenital Anomalies of the Female Reproductive Tract in a Patient with Goltz Syndrome. J Pediatr Adolesc Gynecol. 2009;22(4):e71-72. doipubmed
Lopez-Porras RF, Arroyo C, Soto-Vega E. Focal Dermal Hypoplasia with Uterus Bicornis and Renal Ectopia: Case Report and Review of the Literature. Case Rep Dermatol. 2011;3(2):158-163. doipubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
International Journal of Clinical Pediatrics is published by Elmer Press Inc.