

| International Journal of Clinical Pediatrics, ISSN 1927-1255 print, 1927-1263 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, Int J Clin Pediatr and Elmer Press Inc |
| Journal website http://www.theijcp.org |
Original Article
Volume 4, Number 1, March 2015, pages 121-126
GJB2 Gene Testing, Etiologic Diagnosis and Genetic Counseling in Romanian Persons With Prelingual Hearing Loss
Cristina Dragomira, Adriana Stana, Dragos T. Stefanescua, Codrut Sarafoleanub, Emilia Severinc, d, Lorand Savua
aGenetic Lab, Molecular Genetics, Str. Capitan Aviator Nicolae Drossu, Nr. 9, Sector 1, Bucharest, Romania
bCarol Davila University of Medicine and Pharmacy, ENT Department 37, Dionisie Lupu Street, Bucharest, Romania
cGenetics Department, Carol Davila University of Medicine and Pharmacy, 24, Kiseleff Av., 011346 Bucharest, Romania
dCorresponding Author: Emilia Severin, Genetics Department, “Carol Davila” University of Medicine and Pharmacy, 24, Kiseleff Av., 011346 Bucharest, Romaniaor
Manuscript accepted for publication March 20, 2015
Short title: Testing for Sensorineural Hearing Loss
doi: http://dx.doi.org/10.14740/ijcp194w
| Abstract | ▴Top |
Background: Nowadays, molecular genetic tests provide insights into the etiologic diagnosis of hearing loss (HL). Specific gene mutations are known to cause sensorineural HL of early onset. Previously published studies showed the allelic heterogeneity of GJB2 gene as main genetic cause of isolated congenital HL. The aims of the present study were to provide an extended and updated spectrum of mutations in GJB2 gene and to identify the most prevalent mutations in the Romanian population for testing prevention strategy in people with sensorineural HL of early onset.
Methods: To overcome our aims, we used clinical data from 125 unrelated persons with congenital HL and performed ARMS-PCR and DNA sequencing techniques for detection of known mutations or identification of mutations within GJB2 gene.
Results: The most prevalent mutation was c.35delG found in both homozygotic and heterozygotic forms. The second mutant allele was c.71G>A (p.W24X) found in homo- or heterozygotic forms as well, followed by c.-23+1G>A and c.380G>A (p.R127H) mutations with lower frequencies.
Conclusion: The study reveals the c.35delG mutation as having the highest prevalence, further highlights the genetic background of congenital HL in a local population, and supports improvement of genetic testing such as newborn and carrier screening on which to base genetic counseling services.
Keywords: GJB2 gene sequencing; Congenital hearing loss etiology; Alleles for first-line screening; Genetic counseling
| Introduction | ▴Top |
Hearing loss (HL) is one of the most common health problems worldwide that affects a large number of people of all age groups and both genders (http://www.who.int/mediacentre/factsheets/fs300/en/, accessed January 21, 2015). In society, deaf or hard of hearing persons can function well with the help of new technology, hearing aids, and cochlear implants [1] if they get proper diagnosis and early intervention based on a comprehensive HL evaluation. Non-syndromic HL and deafness is characterized by childhood onset, progressive, moderate-to-severe, high-frequency sensorineural hearing impairment. The audioprofile may vary significantly, even among family members. Affected individuals have no other associated medical findings [2]. At the moment, molecular genetic testing is part of such evaluation detecting specific mutations involved in sensorineural HL of early onset. The c.35delG mutation in the GJB2 gene, which encodes connexin 26, has been reported to be the most prevalent GJB2 mutation among Caucasians. Connexin 26 is expressed with connexin 30 (encoded by the GJB6 gene) in the inner ear. Two deletions in the GJB6 gene, namely del(GJB6-D13S1830) encompassing 309 kb [3-5] and del(GJB6-D13S1854) spanning 232 kb, have been shown to be a common cause of HL as well. The findings of our previous work indicated no detection of deletions in the GJB6 gene in Romanian population [6] and suggested exclusion of GJB6 gene from the present study.
So, previously published study [7] about the c.35delG carrier rate in the Romanian population (estimated to be 3.14%), the literature data about ethnic or geographic distribution and genetic epidemiology of the GJB2 gene and the incidence of congenital HL in Romania (estimated to be 0.93 per 1,000 live births) designed our study. It is known that many different mutations of the GJB2 gene are capable of altering structure or function of connexin 26 protein. The aims of the present study were to provide an extended and updated mutational spectrum in GJB2 gene and to identify the most prevalent mutations in the Romanian population for first-line genetic screening tests.
| Methods | ▴Top |
Clinical findings
We tested 125 unrelated children clinically diagnosed with HL, with an unremarkable family history, from all geographic regions of Romania. The children were ranging in age from 1 to 13 years (at the moment of the study) and presented bilateral HL from mild to profound forms. Written informed consent was obtained from all the parents of minors.
The otolaryngologists working in different medical centers from all over the country performed medical interviews and physical examinations. The subjects were referred to our laboratory since genetic testing could be used to determine (or attempt to determine) the cause of their sensorineural HL. Genetic counseling was offered (by a genetic professional) to all the families and more than half of all parents had a good understanding of genetics of HL.
The sample selection was consecutive and all available patients were included in order to obtain a better representation of the entire Romanian population.
Inclusion criteria were: 1) time/age of onset: congenital - prelingual (before the acquisition of speech); 2) clinical presentation: non-syndromic HL/no related medical findings; 3) type of HL: sensorineural; 4) prognosis: stable/progressive HL; 5) both ears affected; 6) sporadic case in his/her family; and 7) persons with unaffected parents.
Exclusion criteria were: 1) syndromic HL; and 2) congenital HL caused by infections.
The period of study took place between June 2012 and February 2014.
Genomic DNA was extracted from peripheral blood cells with QIAmp DNA Blood Mini Kit (QIAGEN).
ARMS-PCR
To detect c.35delG GJB2 gene mutation, we used ARMS-PCR. The two PCR assays using either the normal or mutant primer along with the common primer proposed by Scott et al (1998) for the GJB2 gene detect the presence of the normal or mutant allele, with the corresponding amplicon size at 202 pb [8]. We introduced β-actin (294 pb) as an internal control to monitor efficiency of both reactions (Fig. 1) [6].
 Click for large image | Figure 1. Detection of c.35delG mutation. (A) PCR amplification of DNA using normal and common primers for GJB2 and β-actin primers. (B) PCR amplification of DNA using mutant and common primers for GJB2 and β-actin primers. Lane A01: pUC18/HaeIII marker; lane A02: homozygous 35delG positive control; lane A03: heterozygous c.35delG positive control; lanes A04 to A06: wild type DNA samples; lane A07: homozygous c.35delG DNA sample; lane A08: wild type DNA sample; lanes A09 to A10: heterozygous c.35delG DNA samples; lane A11: homozygous c.35delG DNA sample; lane A12: negative control (a sample to which no DNA was added during the PCR). |
DNA sequencing
The entire coding exon 2 of GJB2 gene was amplified using the three primer sets and PCR conditions described by Mkaouar-Rebai et al (2007) [9]. The expected sizes of the amplification products were 286, 279 and 279 bp, respectively. The PCR was performed in a final volume of 50 µL containing 200 ng genomic DNA, 10 pmol each primer, 1 × PCR buffer, 1.5 mM MgCl2, 0.2 mM dNTP and 3 U Taq DNA polymerase (Promega).
The non-coding exon 1 was amplified using the primer set described by Ferraris et al (2002) [10]: 5’-TCC GTA ACT TTC CCA TGTC-3’ and 5’-CTC TGG GTC TCG CGG TC-3’.
The amplicon includes the region of the exon 1 where is located the splice sites mutations c.-23+1 G>A originally named IVS1+1 G→A. PCR was performed in 50 µL reactions containing 200 ng genomic DNA, 10 pmol each primer, 1 × PCR buffer, 1.5 mM MgCl2, 0.2 mM each dNTP, and 3 U Taq DNA polymerase (Promega). PCR was carried out using the following program: initial denaturation at 95 °C for 5 min, five touchdown cycles of denaturation at 94 °C for 1 min, annealing for 50 s at 63 °C for the first cycle and 1 °C reduction per cycle, extension 1 min at 72 °C, and then 40 cycles of denaturation at 94 °C for 1 min, annealing at 59 °C for 50 s and extension at 72 °C for 1 min. Final extension was at 72 °C for 7 min. The PCR reactions were performed on a GeneAmp PCR System 9700 thermal cycler (Applied Biosystem). After purification of the PCR product (Wizard SV Gel and PCR Clean-up System, Promega), bi-directional DNA sequencing with the PCR primers on CEQ8800 Genetic Analyzer (Beckman Coulter) was performed. The blast homology searches were performed using the programs available at the UCSC Genome Bioinformatics Website compared with the wild type GJB2 gene sequence.
Ethical approval
The Research Ethics Committee of the Carol Davila University of Medicine and Pharmacy reviewed and approved the study protocol (CERO - CAREER PROFILE: ROMANIAN RESEARCHER, POSDRU/159/1.5/S/135760), which conformed to the ethical guidelines of the Declaration of Helsinki, on the principles for medical research involving human subjects.
| Results | ▴Top |
Using ARMS we analyzed all 125 DNA samples and detected 46 children homozygous for c.35del mutation. Forty-two homozygous children for c.35delG mutation presented profound bilateral HL, two had severe, one moderate and one mild impairment according to clinical data (Table 1). The results show the c.35delG mutation to be present in the homozygous state (46/125 or 36.8%) and heterozygous state (11/125 or 8.8%) as well. GJB2 gene sequencing was used to analyze the remaining 79 DNA samples including the 68 children who were previously detected negative for the c.35delG mutation and the 11 children found heterozygous for the c.35delG mutation. Both the coding region of exon 2 and the region of splice site of non-coding exon 1 of the GJB2 gene were sequenced (Fig. 2). After sequencing of the GJB2 gene we found the c.71G>A (p.W24X), c.-23+1G>A and c.380G>A (p.R127H) mutations. All results are presented in Table 2.
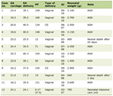 Click to view | Table 1. Clinical Manifestations of the c.35delG Homozygous Individuals With HL |
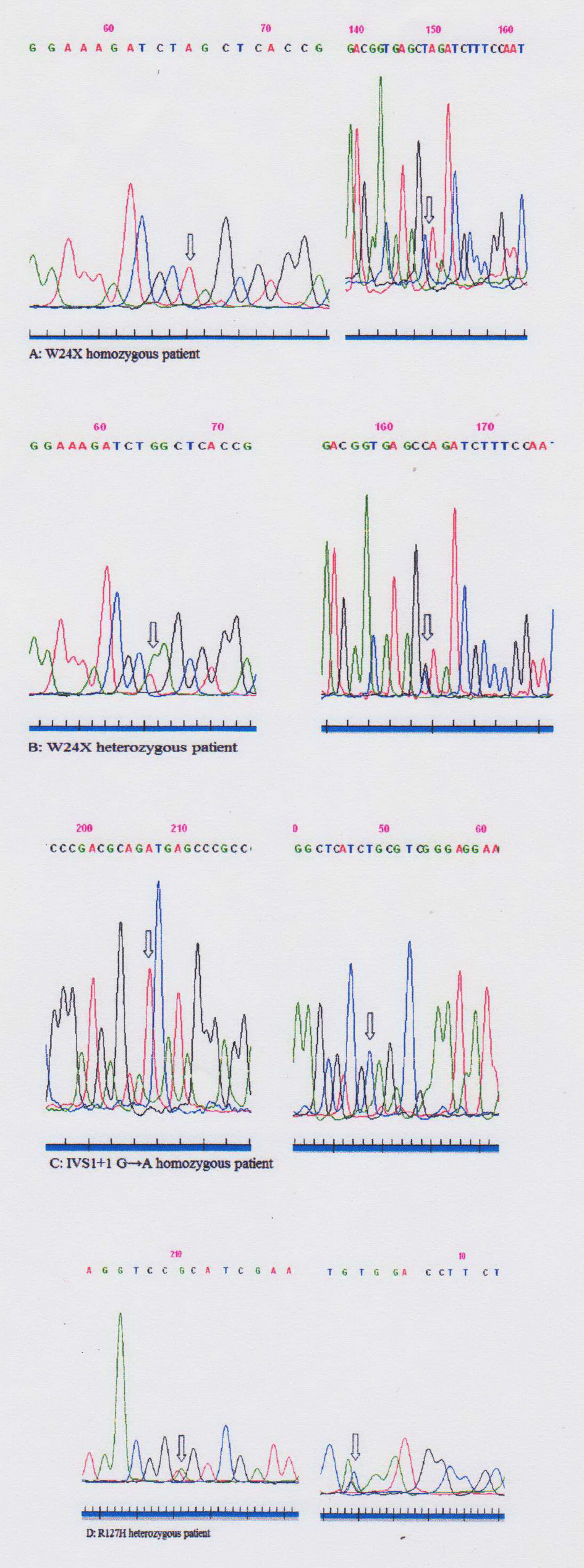 Click for large image | Figure 2. Sequences of GJB2 gene fragments of patients (A-D). Sequencing with forward (left) and reverse (right) primers. Mutations are highlighted with arrows. |
Click to view | Table 2. List of Identified Mutations After the Sequencing of the GJB2 Identified in the Hearing Impaired Romanian Children |
| Discussion | ▴Top |
GJB2 is the most widely sequenced HL gene and over 100 mutations of this gene are associated with HL. In addition, our data re-confirm presence of p.W24X (c.71G>A) mutation in Romanian population. This mutation was also found in North-West Romania with a frequency rate of 5.3% among the children with non-syndromic hearing loss (NSHL) [11]. The mutation was first described in two Pakistani families [12]. Other studies show a high frequency of p.W24X mutation in several Asian families [13, 14]. This mutation was also found to be a common allele in Spanish Romany populations as well [15]. The p.W24X mutation was also identified in Slovak Romany families [16].
Most patients found homozygous for the c.35delG or p.W24X mutations and compound heterozygous for c.35delG/W24X mutations presented profound forms of HL (Table 1, 2). The c.35delG and p.W24X are known as truncating mutations. The c.35delG mutation, also known as 30delG, is predicted to inactivate connexin 26. The single base deletion within a stretch of six Gs between position +30 and +35 results in a frameshift leading to a UGA stop codon, two residues downstream (codon 13) and premature termination of the connexin 26 synthesis at the 12th amino acid [17]. The c.71G>A (p.W24X) mutation is a nonsense mutation consisting of a G-to-A transition at position 71, resulting in a trp24-to-ter substitution (24 amino acids instead of 226 amino acid). In a homozygous state, no functional monomers are present in the cells [12]. However, within the group of patients with a homozygous c.35delG genotype variations were observed with the HL ranging from mild or moderate (least often) to severe and profound (most frequent). This phenotypic variation was explained by the influence of modifier genes and/or environmental factors that lead to incomplete penetrance and variable expression [18, 19]. Other studies consider that the phenotypic variation may be caused by a smaller effect of different interacting genes [20].
One relevant finding is the existence of c.-23+1G>A (a splice-site mutation in the non-coding exon 1 of Cx26 at position -3172) among hearing impaired Romanian children. The allele frequency was found between 1% and 9.4% for GJB2 mutations among the Asian and Brazilian populations [21-23] and much higher in the Hungarian and Czech populations, around 23.4% and 45%, respectively [24, 25]. This mutation is predicted to disrupt splicing, yielding no detectable mRNA [26] and is consistent with our data given that both patients presented profound bilateral hearing impairment.
We also identified a heterozygous p.R127H (c.380G>A) mutation in two children with mild HL without any additional mutations in GJB2. This mutation consisting of a G to A substitution at position 380, codon 127, leads to the replacement of an arginine with a histidine. The mutation was previously described in hearing impaired patients as the sole mutation in GJB2 gene, affecting a residue that is not highly conserved suggesting nonpathogenic nature of this mutation [27, 28]. This mutation is considered as a polymorphism in some studies [29-33], while in other studies is considered to affect function of gap junction channels at different levels of protein expression [27, 34, 35].
Analysis of the GJB2 gene in a total of 125 HL cases revealed that 46.4% of hearing impaired children did not have any mutation in the GJB2 gene. At this time, genetic tests for most common mutations associated with HL are available. We should analyze additional genes responsible for recessive HL other than DFNB1 (GJB2 and GJB6) in the negative patients in the future.
Conclusions
In summary, variants of the GJB2 gene of known clinical significance were found in Romanians with congenital NSHL. The most prevalent mutation is c.35delG, similarly to other European populations. The patterns of associated alleles have different frequencies compared with neighboring countries and characterize the genetic background of Romanian population.
The results have also biomedical implications. The genetic testing and counseling integrated into clinical care reduce the need to perform further clinical tests for congenital HL. Allelic heterogeneity of GJB2 gene is important for medical genetics services because it applies to NSHL as a Mendelian disorder and defines new guidelines for clinical practice as well. Due to the allelic heterogeneity [36], there are a large number of normal hearing heterozygotes (carrier screening) but identification of prevalent mutations allows for prenatal and family-specific carrier screening. Genetic counseling should be offered in case of marriages between individuals with GJB2 mutations in their families as an opportunity for prevention.
Acknowledgement
The authors thank to all patients and their families for their participation in the study. The study was supported by project CERO - CAREER PROFILE: ROMANIAN RESEARCHER, POSDRU/159/1.5/S/135760.
Conflict of Interest
None of the authors has any conflict of interest, financial or otherwise.
| References | ▴Top |
- World Health Organization (WHO). http://www.who.int/mediacentre/factsheets/fs300/en/. Accessed January 21, 2015.
- Smith RJH, Shearer AE, Hildebrand MS, Van Camp G: Deafness and Hereditary Hearing Loss Overview. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, et al., eds. GeneReviews(R). Seattle (WA), 1993.
- Lerer I, Sagi M, Ben-Neriah Z, Wang T, Levi H, Abeliovich D. A deletion mutation in GJB6 cooperating with a GJB2 mutation in trans in non-syndromic deafness: A novel founder mutation in Ashkenazi Jews. Hum Mutat. 2001;18(5):460.
doi pubmed - del Castillo I, Villamar M, Moreno-Pelayo MA, del Castillo FJ, Alvarez A, Telleria D, Menendez I, et al. A deletion involving the connexin 30 gene in nonsyndromic hearing impairment. N Engl J Med. 2002;346(4):243-249.
doi pubmed - Del Castillo I, Moreno-Pelayo MA, Del Castillo FJ, Brownstein Z, Marlin S, Adina Q, Cockburn DJ, et al. Prevalence and evolutionary origins of the del(GJB6-D13S1830) mutation in the DFNB1 locus in hearing-impaired subjects: a multicenter study. Am J Hum Genet. 2003;73(6):1452-1458.
doi pubmed - Dragomir C, Stan A, Stefanescu DT, Savu L, Severin E. Prenatal screening for the 35delG GJB2, del (GJB6-D13S1830), and del (GJB6-D13S1854) mutations in the Romanian population. Genet Test Mol Biomarkers. 2011;15(11):749-753.
doi pubmed - Dragomir C, Stan A, Stefanescu DT, et al. Confirmation of diagnosis in children with DFNB1 related deafness. American Journal of Molecular Biology. 2013;3(4): 229-234.
doi - Scott DA, Kraft ML, Carmi R, et al. Identification of mutations in the connexin 26 gene that cause autosomal recessive non-syndromic hearing loss. Hum Mutation. 1998;11:487-494.
doi - Mkaouar-Rebai E, Tlili A, Masmoudi S, Belguith N, Charfeddine I, Mnif M, Triki C, et al. Mutational analysis of the mitochondrial tRNALeu(UUR) gene in Tunisian patients with mitochondrial diseases. Biochem Biophys Res Commun. 2007;355(4):1031-1037.
doi pubmed - Ferraris A, Rappaport E, Santacroce R, Pollak E, Krantz I, Toth S, Lysholm F, et al. Pyrosequencing for detection of mutations in the connexin 26 (GJB2) and mitochondrial 12S RNA (MTRNR1) genes associated with hereditary hearing loss. Hum Mutat. 2002;20(4):312-320.
doi pubmed - Lazar C, Popp R, Trifa A, Mocanu C, Mihut G, Al-Khzouz C, Tomescu E, et al. Prevalence of the c.35delG and p.W24X mutations in the GJB2 gene in patients with nonsyndromic hearing loss from North-West Romania. Int J Pediatr Otorhinolaryngol. 2010;74(4):351-355.
doi pubmed - Kelsell DP, Dunlop J, Stevens HP, Lench NJ, Liang JN, Parry G, Mueller RF, et al. Connexin 26 mutations in hereditary non-syndromic sensorineural deafness. Nature. 1997;387(6628):80-83.
doi pubmed - Kudo T, Ikeda K, Kure S, Matsubara Y, Oshima T, Watanabe K, Kawase T, et al. Novel mutations in the connexin 26 gene (GJB2) responsible for childhood deafness in the Japanese population. Am J Med Genet. 2000;90(2):141-145.
doi - Maheshwari M, Vijaya R, Ghosh M, Shastri S, Kabra M, Menon PS. Screening of families with autosomal recessive non-syndromic hearing impairment (ARNSHI) for mutations in GJB2 gene: Indian scenario. Am J Med Genet A. 2003;120A(2):180-184.
doi pubmed - Alvarez A, del Castillo I, Villamar M, Aguirre LA, Gonzalez-Neira A, Lopez-Nevot A, Moreno-Pelayo MA, et al. High prevalence of the W24X mutation in the gene encoding connexin-26 (GJB2) in Spanish Romani (gypsies) with autosomal recessive non-syndromic hearing loss. Am J Med Genet A. 2005;137A(3):255-258.
doi pubmed - Minarik G, Tretinarova D, Szemes T, Kadasi L. Prevalence of DFNB1 mutations in Slovak patients with non-syndromic hearing loss. Int J Pediatr Otorhinolaryngol. 2012;76(3):400-403.
doi pubmed - Zelante L, Gasparini P, Estivill X, Melchionda S, D'Agruma L, Govea N, Mila M, et al. Connexin26 mutations associated with the most common form of non-syndromic neurosensory autosomal recessive deafness (DFNB1) in Mediterraneans. Hum Mol Genet. 1997;6(9):1605-1609.
doi pubmed - Nadeau JH. Modifier genes in mice and humans. Nat Rev Genet. 2001;2(3):165-174.
doi pubmed - Snoeckx RL, Huygen PL, Feldmann D, Marlin S, Denoyelle F, Waligora J, Mueller-Malesinska M, et al. GJB2 mutations and degree of hearing loss: a multicenter study. Am J Hum Genet. 2005;77(6):945-957.
doi pubmed - Hilgert N, Huentelman MJ, Thorburn AQ, Fransen E, Dieltjens N, Mueller-Malesinska M, Pollak A, et al. Phenotypic variability of patients homozygous for the GJB2 mutation 35delG cannot be explained by the influence of one major modifier gene. Eur J Hum Genet. 2009;17(4):517-524.
doi pubmed - Mahdieh N, Rabbani B, Wiley S, Akbari MT, Zeinali S. Genetic causes of nonsyndromic hearing loss in Iran in comparison with other populations. J Hum Genet. 2010;55(10):639-648.
doi pubmed - da Silva-Costa SM, Coeli FB, Lincoln-de-Carvalho CR, Marques-de-Faria AP, Kurc M, Pereira T, Pomilio MC, et al. Screening for the GJB2 c.-3170 G>A (IVS 1+1 G>A) mutation in Brazilian deaf individuals using multiplex ligation-dependent probe amplification. Genet Test Mol Biomarkers. 2009;13(5):701-704.
doi pubmed - Tekin M, Xia XJ, Erdenetungalag R, Cengiz FB, White TW, Radnaabazar J, Dangaasuren B, et al. GJB2 mutations in Mongolia: complex alleles, low frequency, and reduced fitness of the deaf. Ann Hum Genet. 2010;74(2):155-164.
doi pubmed - Seeman P, Sakmaryova I. High prevalence of the IVS 1 + 1 G to A/GJB2 mutation among Czech hearing impaired patients with monoallelic mutation in the coding region of GJB2. Clin Genet. 2006;69(5):410-413.
doi pubmed - Toth T, Kupka S, Haack B, Fazakas F, Muszbek L, Blin N, Pfister M, et al. Coincidence of mutations in different connexin genes in Hungarian patients. Int J Mol Med. 2007;20(3):315-321.
doi - Shahin H, Walsh T, Sobe T, Lynch E, King MC, Avraham KB, Kanaan M. Genetics of congenital deafness in the Palestinian population: multiple connexin 26 alleles with shared origins in the Middle East. Hum Genet. 2002;110(3):284-289.
doi pubmed - Estivill X, Fortina P, Surrey S, Rabionet R, Melchionda S, D'Agruma L, Mansfield E, et al. Connexin-26 mutations in sporadic and inherited sensorineural deafness. Lancet. 1998;351(9100):394-398.
doi - Rabionet R, Zelante L, Lopez-Bigas N, D'Agruma L, Melchionda S, Restagno G, Arbones ML, et al. Molecular basis of childhood deafness resulting from mutations in the GJB2 (connexin 26) gene. Hum Genet. 2000;106(1):40-44.
doi pubmed - Marlin S, Garabedian EN, Roger G, Moatti L, Matha N, Lewin P, Petit C, et al. Connexin 26 gene mutations in congenitally deaf children: pitfalls for genetic counseling. Arch Otolaryngol Head Neck Surg. 2001;127(8):927-933.
doi pubmed - RamShankar M, Girirajan S, Dagan O, Ravi Shankar HM, Jalvi R, Rangasayee R, Avraham KB, et al. Contribution of connexin26 (GJB2) mutations and founder effect to non-syndromic hearing loss in India. J Med Genet. 2003;40(5):e68.
doi pubmed - Roux AF, Pallares-Ruiz N, Vielle A, Faugere V, Templin C, Leprevost D, Artieres F, et al. Molecular epidemiology of DFNB1 deafness in France. BMC Med Genet. 2004;5:5.
doi pubmed - Thonnissen E, Rabionet R, Arbones ML, Estivill X, Willecke K, Ott T. Human connexin26 (GJB2) deafness mutations affect the function of gap junction channels at different levels of protein expression. Hum Genet. 2002;111(2):190-197.
doi pubmed - Wang HL, Chang WT, Li AH, Yeh TH, Wu CY, Chen MS, Huang PC. Functional analysis of connexin-26 mutants associated with hereditary recessive deafness. J Neurochem. 2003;84(4):735-742.
doi pubmed - D'Andrea P, Veronesi V, Bicego M, Melchionda S, Zelante L, Di Iorio E, Bruzzone R, et al. Hearing loss: frequency and functional studies of the most common connexin26 alleles. Biochem Biophys Res Commun. 2002;296(3):685-691.
doi - Palmada M, Schmalisch K, Bohmer C, Schug N, Pfister M, Lang F, Blin N. Loss of function mutations of the GJB2 gene detected in patients with DFNB1-associated hearing impairment. Neurobiol Dis. 2006;22(1):112-118.
doi pubmed - Gandia M, Del Castillo FJ, Rodriguez-Alvarez FJ, Garrido G, Villamar M, Calderon M, Moreno-Pelayo MA, et al. A novel splice-site mutation in the GJB2 gene causing mild postlingual hearing impairment. PLoS One. 2013;8(9):e73566.
doi pubmed
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
International Journal of Clinical Pediatrics is published by Elmer Press Inc.