

| International Journal of Clinical Pediatrics, ISSN 1927-1255 print, 1927-1263 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, Int J Clin Pediatr and Elmer Press Inc |
| Journal website http://www.theijcp.org |
Case Report
Volume 6, Number 3-4, December 2017, pages 51-53
Status Dystonicus: A Rare Presentation of Molybdenum Cofactor Deficiency
Kursat Bora Carmana, d, Gonca Kilic Yildirimb, Eylem Kiralc, Ozan Kocaka, Sibel Lacinel Gurlevika, Coskun Yarara, Ener Cagri Dinleyicic, Sultan Durmus Aydogdub
aDepartment of Pediatric Neurology, Eskisehir Osmangazi University Hospital, Eskisehir, Turkey
bDepartment of Pediatric Metabolic Diseases, Eskisehir Osmangazi University Hospital, Eskisehir, Turkey
cDepartment of Pediatric Intensive Care, Eskisehir Osmangazi University Hospital, Eskisehir, Turkey
dCorresponding Author: Kursat Bora Carman, Department of Pediatric Neurology, Eskisehir Osmangazi University Hospital, Eskisehir, Turkey
Manuscript submitted October 30, 2017, accepted December 12, 2017
Short title: Status Dystonicus and MOCOD
doi: https://doi.org/10.14740/ijcp283w
| Abstract | ▴Top |
Molybdenum cofactor deficiency (MOCOD) is a severe inborn error of metabolism. It is characterized by neonatal presentation of intractable seizures, feeding difficulties, severe developmental delay, microcephaly with brain atrophy and coarse facial features. Movement disorders have been rarely reported as presenting complaint of MOCOD. We report a previously healthy 10 months old boy presenting with acute onset status dystonicus. A novel homozygote IVS1-1G>A (c.251-1G>A) mutation was determined and he was diagnosed as MOCOD. MOCOD must be considered in the differential diagnosis of movement disorders.
Keywords: Status dystonicus; Children; Molybdenum cofactor; Metabolism
| Introduction | ▴Top |
Molybdenum cofactor deficiency (MOCOD, OMIM: 252150) is a rare autosomal recessive neurometabolic disease [1, 2]. The functions of three enzymes, sulfite oxidase, xanthine dehydrogenase and aldehyde oxidase altered. Patients typically present with neonatal seizure, severe neurodevelopmental delay, epileptic encephalopathy, lens dislocation, feeding difficulties and dysmorphias [3]. Movement disorders have been rarely reported as presenting complaint of MOCOD [4-6].
We report a child presenting with status dystonicus, who was diagnosed as MOCOD based on low level of serum uric acid and gene analysis.
| Case Report | ▴Top |
A 10-month-old boy referred to emergency unit because of sudden onset opisthotonic posture. He was the only child of non-consanguineous parents. The mother and father were 30 years old and 33 years old respectively and both are officers. There was no family history of genetic illness. His parents reported that he was previously healthy and had fever and cough for 2 days. No medication was used to treat those symptoms. On the day of presentation while he was playing with toys, he developed opisthotonos suddenly. He was born by cesarean section after a normal pregnancy at 38 weeks of gestation, and was the first child. His birth weight was 3,200 g. His developmental milestones were reported as appropriated to his age by parents. However previous medical reports of him could not be obtained to confirm that neurodevelopment of his was appropriate.
A physical and neurologic examination revealed a head circumference of 42 cm, a height of 64 cm, and a weight of 5,860 g, all of which were at or below the 5th percentile for his age. He was on opisthotonic posture (Fig. 1a). He had generalized increase in muscle tone. Deep tendon reflexes were hyperactive. Enteral feeding was impossible. He was hospitalized in intensive care unit and treatment with diazepam (0.3 mg/kg) and baclofen 2 × 2.5 mg was started. No clinical response was observed. Midazolam infusion (0.2 mg/kg/h) was added to the treatment. Because of unresponsiveness, the dose of baclofen was increased (4 mg/kg/d). Status dystonicus subsided and the patient was relaxed (Fig. 1b). The results of routine laboratory investigations of blood and urine involving serum lactate, pyruvate and ammonia, urine and plasma amino acids concentrations were all within normal limits except serum uric acid level (0.3 mg/dL). The repeated uric acid levels were also low. There was increased urinary excretion of sulfite (dipstick test positive). Brain magnetic resonance imaging showed corpus callosum atrophy, enlargement of lateral ventricular dilatation, and multicystic encephalomalacia. The results of ophthalmologic, echocardiographic, abdominal ultrasonographic examinations were all normal. Electroencephalography (EEG) revealed normal sleep pattern and no epileptiform discharges. To confirm, the MOCOD diagnosis, genetic analysis was performed. Sequencing MOCS1 gene revealed previously unreported homozygote IVS1-1G>A (c.251-1G>A) mutation in the sixth chromosome, p21.2 region (Fig. 2). He was discharged on oral baclofen treatment. The patient moved to another city and it was learned that he died 3 months later.
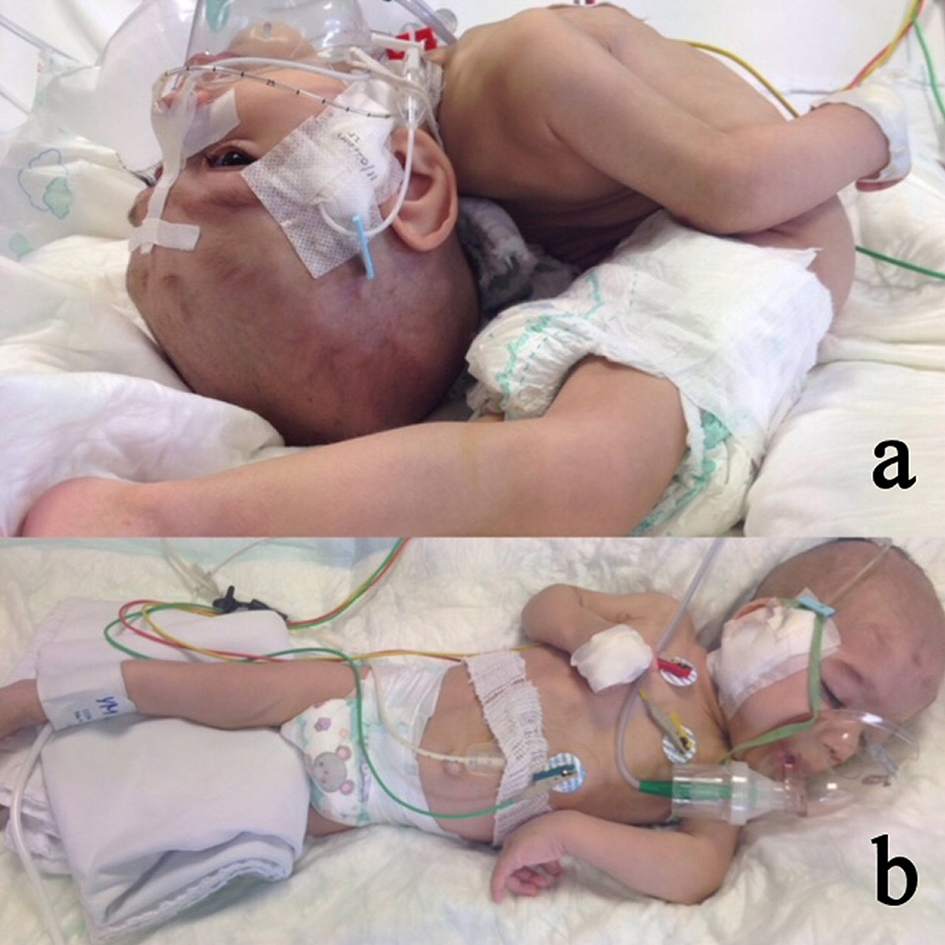 Click for large image | Figure 1. The posture of the patient before (a) and after the treatment (b). |
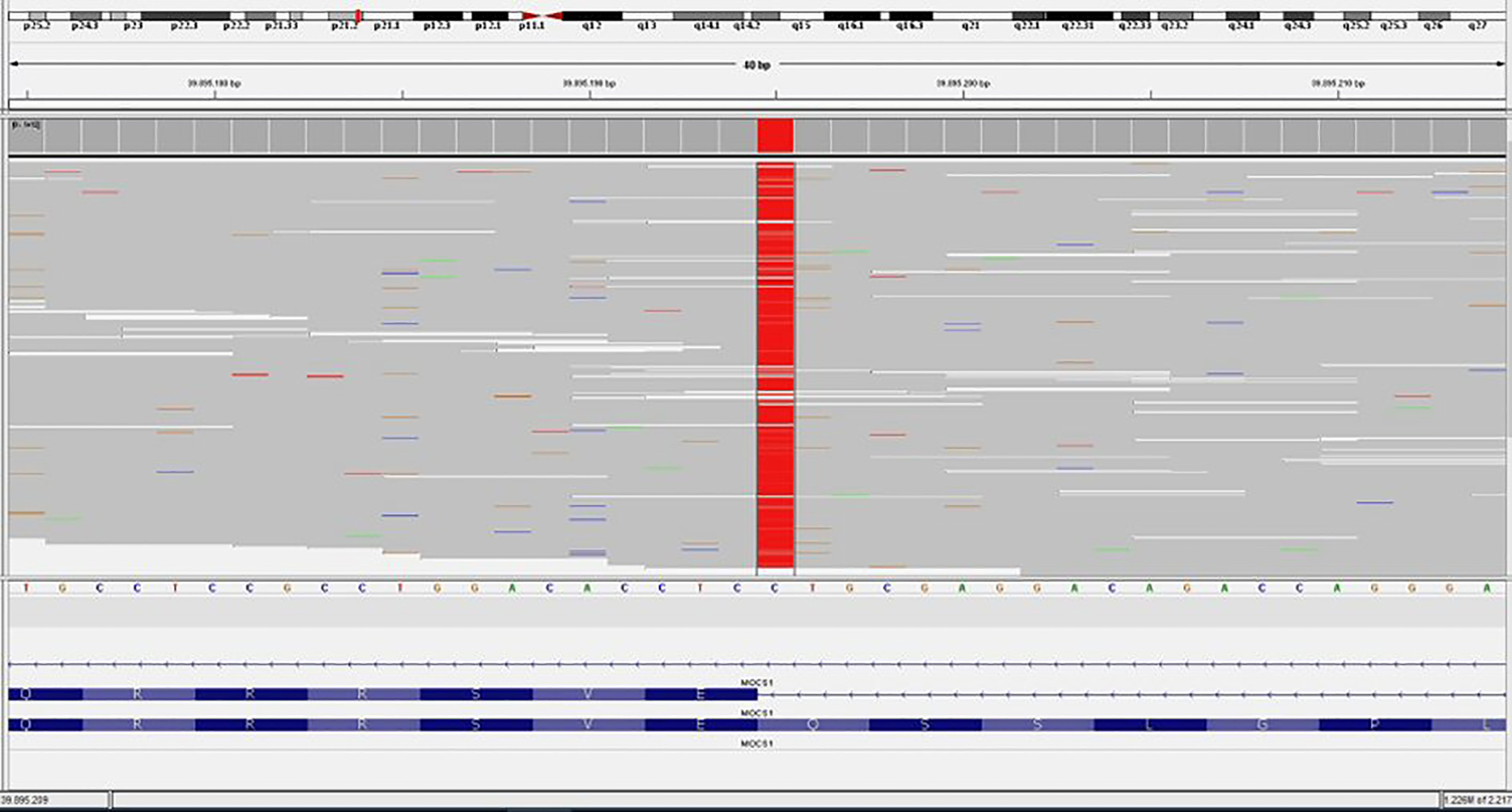 Click for large image | Figure 2. Illustration of DNA sequencing of patient. |
A written consent was received from the parents of the patient.
| Discussion | ▴Top |
In the patient described here, movement disorder “status dystonicus” was the presenting complaint of MOCOD. Movement disorders are rarely chief complaint of MOCOD. Ichida et al [4] reported a 7-year-old boy presenting with mental retardation, dystonic movements and seizure. That patient has had also mutation on MOCS1 gene like our case. A 2-year-old boy presented with spastic quadriplegia who was previously misdiagnosed as cerebral palsy, and sequencing of the MOCS2A gene revealed heterozygosity for (c.265T>C) + (266A>G), which was diagnosed as MOCODB [5]. However, no phenotype genotype correlation is present. A 2-year-old patient was described presenting with spasticity and psychomotor retardation and a homozygous c.1739A>C transversion in exon 18 was reported.
MOCOD might present with parkinsonian features. Alkufri et al [6] reported an adult with progressive apathy, slowness of movements, tremor and generalized parkinsonism was reported by and diagnosed as MOCOD.
Status dystonicus is a life-threatening movement disorder emergency [7]. It is often a triggered event. The parents of our case reported that the patient was previously healthy and had fever and cough for 2 days and onset of status dystonicus was sudden. Infection, drugs, trauma, surgery, “metabolic disorder” decompensation, stress and pain might trigger status dystonicus [8, 9]. It is possible that infection and fever were triggers of present case.
Many different drugs, used individually or in combination, can be used to treat status dystonicus including baclofen, benzodiazepines, L-3,4-dihydroxyphenylalanine (L-DOPA), tetrabenazine, benzhexol, choral hydrate, propofol, barbiturates, pimozide, haloperidol and anti-cholinergics with varying success rates. The non-responded patients may require more invasive treatments such as intrathecal baclofen, deep brain stimulation or neurosurgery [8-11]. The pediatric dose of oral baclofen is uncertain [12, 13]. In our case, pharmacological therapy by baclofen was (4 mg/kg/day) successful and our patient improved. There is no definitive treatment of MOCOD [3, 14]. The present case was not followed and died within 3 months.
In conclusion, the present case demonstrated that a patient with status dystonicus of underdetermined cause should undergo testing for uric acid in both serum and urine. Low serum uric acid level and genetic analysis may lead to a correct diagnosis of MOCOD.
Conflict of Interest
The authors declare no conflict of interest.
Financial Support
The authors declare no financial assistance.
| References | ▴Top |
- Schwarz G, Mendel RR, Ribbe MW. Molybdenum cofactors, enzymes and pathways. Nature. 2009;460(7257):839-847.
doi pubmed - Duran M, Beemer FA, van de Heiden C, Korteland J, de Bree PK, Brink M, Wadman SK, et al. Combined deficiency of xanthine oxidase and sulphite oxidase: a defect of molybdenum metabolism or transport? J Inherit Metab Dis. 1978;1(4):175-178.
doi pubmed - Atwal PS, Scaglia F. Molybdenum cofactor deficiency. Mol Genet Metab. 2016;117(1):1-4.
doi pubmed - Ichida K, Aydin HI, Hosoyamada M, Kalkanoglu HS, Dursun A, Ohno I, Coskun T, et al. A Turkish case with molybdenum cofactor deficiency. Nucleosides Nucleotides Nucleic Acids. 2006;25(9-11):1087-1091.
doi pubmed - Kikuchi K, Hamano S, Mochizuki H, Ichida K, Ida H. Molybdenum cofactor deficiency mimics cerebral palsy: differentiating factors for diagnosis. Pediatr Neurol. 2012;47(2):147-149.
doi pubmed - Alkufri F, Harrower T, Rahman Y, Hughes E, Mundy H, Knibb JA, Moriarty J, et al. Molybdenum cofactor deficiency presenting with a parkinsonism-dystonia syndrome. Mov Disord. 2013;28(3):399-401.
doi pubmed - Singer HS, Mink JW, Gilbert DL, Jankovic J. Dystonia. Movement disorders in childhood,1st ed. Philadelphia, Elsevier Saunders; 2010. p. 97-109.
doi - Allen NM, Lin JP, Lynch T, King MD. Status dystonicus: a practice guide. Dev Med Child Neurol. 2014;56(2):105-112.
doi pubmed - Grosso S, Verrotti A, Messina M, Sacchini M, Balestri P. Management of status dystonicus in children. Cases report and review. Eur J Paediatr Neurol. 2012;16(4):390-395.
doi pubmed - Kyriagis M, Grattan-Smith P, Scheinberg A, Teo C, Nakaji N, Waugh M. Status dystonicus and Hallervorden-Spatz disease: treatment with intrathecal baclofen and pallidotomy. J Paediatr Child Health. 2004;40(5-6):322-325.
doi pubmed - Walcott BP, Nahed BV, Kahle KT, Duhaime AC, Sharma N, Eskandar EN. Deep brain stimulation for medically refractory life-threatening status dystonicus in children. J Neurosurg Pediatr. 2012;9(1):99-102.
doi pubmed - Vaamonde J, Narbona J, Weiser R, Garcia MA, Brannan T, Obeso JA. Dystonic storms: a practical management problem. Clin Neuropharmacol. 1994;17(4):344-347.
doi pubmed - He Y, Brunstrom-Hernandez JE, Thio LL, Lackey S, Gaebler-Spira D, Kuroda MM, Stashinko E, et al. Population pharmacokinetics of oral baclofen in pediatric patients with cerebral palsy. J Pediatr. 2014;164(5):1181-1188 e1188.
- Schwahn BC, Van Spronsen FJ, Belaidi AA, Bowhay S, Christodoulou J, Derks TG, Hennermann JB, et al. Efficacy and safety of cyclic pyranopterin monophosphate substitution in severe molybdenum cofactor deficiency type A: a prospective cohort study. Lancet. 2015;386(10007):1955-1963.
doi
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
International Journal of Clinical Pediatrics is published by Elmer Press Inc.