

| International Journal of Clinical Pediatrics, ISSN 1927-1255 print, 1927-1263 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, Int J Clin Pediatr and Elmer Press Inc |
| Journal website http://www.theijcp.org |
Case Report
Volume 9, Number 1, March 2020, pages 16-19
Asymptomatic Bernard-Soulier Syndrome With a Novel Mutation
Mahmoud Galal Ahmeda, Muzammil Hafeezb, d, Anwar Khanb, Mustafa El Bolkinib, Niyas Parammal Ambadib, Asim Noor Ranac
aDubai Medical College, Dubai, UAE
bNICU, Dubai Hospital, Dubai, UAE
cDepartment of Pediatrics, Dubai Hospital, Dubai, UAE
dCorresponding Author: Muzammil Hafeez, Department of Pediatrics, NICU, PO Box 7272, Dubai Hospital, Dubai, UAE
Manuscript submitted September 4, 2019, accepted November 4, 2019
Short title: Asymptomatic BSS With a Novel Mutation
doi: https://doi.org/10.14740/ijcp342
| Abstract | ▴Top |
Bernard-Soulier syndrome (BSS) is inherited as an autosomal recessive genetic disorder. It is characterized with thrombocytopenia and giant platelets; and various degrees of BSS can be caused by a number of homozygous or compound heterozygous mutations of genes coding for components of the platelet receptor GPIbIX. Here we present a novel mutation occurring in a family causing BSS without any symptoms.
Keywords: Bernard-Soulier syndrome; Novel mutation; GP1BB gene; Asymptomatic
| Introduction | ▴Top |
Bernard-Soulier syndrome (BSS) is inherited as an autosomal recessive genetic disorder. It is a rare disorder, occurring 1 in 1 million. It is characterized by thrombocytopenia, abnormally large platelets, defective adhesion of these platelets to the subendothelium and prolonged bleeding time [1]. BSS is caused by homozygous or compound heterozygous mutations of genes coding for components of the platelet receptor GPIb-IX-V [2]. The GPIb-IX-V, platelet membrane glycoprotein complex, is essential for platelet adhesion at sites of vascular injury through binding to von Willebrand factor (vWF) [3]. The quantitative or qualitative deficiencies in the GPIb-IX-V complex give rise to BSS [4]. The platelets in BSS do not aggregate in response to ristocetin [5]. BSS is clinically characterized by a prolonged bleeding time, and can present as epistaxis, bleeding with minor trauma, ecchymosis without any trauma, bleeding with surgical procedures and sometimes life-threatening bleeding including brain hemorrhage [6]. The platelet count and the genetic defects may not correlate with propensity to bleed and severity of bleeding [6].
Here we present an infant diagnosed as BSS with a novel mutation. This mutation was also found in other members of the family. By knowing the mutations we may be able to correlate with the phenotype and help in the management in future.
| Case Report | ▴Top |
A female infant was born to a 29-year-old mother by normal vaginal delivery at 39 weeks of gestation. Mother had uneventful pregnancy. Her human immunodeficiency virus (HIV), hepatitis B surface antigen (HBsAg), venereal disease research laboratory (VDRL) test, screening for group B streptococcus (GBS) were negative, and she was rubella immune. All her antenatal ultrasound scans were normal.
Parents are non-consanguineous. Father and the elder sibling have low platelet counts. Father’s platelet counts were 38,000/µL and 17,000/µL, respectively on two different occasions. The elder sibling’s platelet count was between 50,000/µL to 110,000/µL. There is no history of bleeding in either of her father or her elder sibling. Blood film showed giant platelets. No other investigations were done for the low platelet count. Mother’s platelet count was 315,000/µL. There was no history of any genetic disorder in the family.
The infant’s birth weight was 2.954 kg, and Apgar score was 8 and 9 at 1 min and 5 min, respectively. Her head circumference was 34 cm, at the 10th centile. She was well perfused and her temperature was 36.5 °C. Her vital signs showed that heart rate of 130 -142/minute, respiratory rate of 40/minute, oxygen saturation of 95-99% in room air, blood pressure 56/35 mm Hg. Her anterior fontanel was normal, systemic examination revealed a normal cardiovascular examination, normal air entry, normal newborn reflexes, no neurological deficits and a normal abdominal examination. She was feeding well from her mother in the postnatal ward. On the fourth day of life, she developed jaundice with bilirubin of 15 mg/dL. Her glucose-6-phosphate dehydrogenase (G6PD) assay was normal. Her mother’s blood group was AB-positive and her blood group was B-positive; and her Coombs test was negative. She was treated with phototherapy and her jaundice resolved. Her full blood count showed a platelet count of 38,000/µL. In view of the thrombocytopenia she was admitted to the neonatal intensive care unit (NICU) and investigated further. Her serial platelet counts were 38,000, 41,000, 51,000, 15,000, 45,000 and 56,000/µL, the blood film showed reduced number of platelets with large and giant platelets (Table 1). Opinion of the pediatric hematologist was taken. In view of the positive family history of thrombocytopenia with giant platelets a provisional diagnosis of Bernard Soulier was made. Bleeding time or coagulation profile was not done. The infant did not develop any petechiae during the stay in NICU and did not require any treatment for the thrombocytopenia. She was discharged from the NICU after observation. The diagnosis of Bernard Soulier was confirmed with whole exome sequence. The whole exome sequence was done for the entire family, which showed that she, her elder sibling and her father are homozygous for the 6-bp duplication c.511_516dup p.(Leu171_Leu172dup) in exon 2 of GP1BB gene (Table 2). Her mother is heterozygous for the same mutation. To the best of knowledge, this variant has neither been described in literature, nor been annotated in genetic data base.
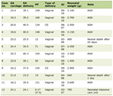 Click to view | Table 1. Laboratory Data on Admission and on Follow-Up |
Click to view | Table 2. Result of Whole Exome Sequence |
The infant was followed in the outpatient clinic. She has normal development milestones. At the age of 3 months she had head control; at 7 months she sat unsupported; at 1 year 3 months she was walking without support, speaking “mama” and “baba”, waving “bye-bye”. Her cardiovascular, neurological and abdominal examination was normal.
| Discussion | ▴Top |
In our patient diagnosed as having BSS, we describe a 6-bp duplication in the exon 2 of the GP1BB gene. The baby was found to have thrombocytopenia on routine blood count at birth; on further investigations, the platelets were abnormally large. BSS is characterized by thrombocytopenia, very large platelets on blood film, prolonged bleeding time and defective adhesion of the platelets to the subendothelium [7]. The characteristic abnormality in BSS is an isolated defect in ristocetin-induced agglutination. Unlike the defect in von Willebrand disease, this abnormality is not corrected by the addition of normal plasma.
Platelet aggregation in response to other agonists, such as collagen and adenosine diphosphate (ADP), as well as clot retraction, is usually normal [8]. In our baby the bleeding time and platelet response to ristocetin were not done due to strong suspicion of BSS; and whole exome sequencing was done. There are about100 described cases of BSS worldwide [1]. Genetic defects have been identified on the GPIba, GPIbβ, and GPIX genes but not on the GPV [8]. In several families, similar mutations in the GPIX have been described. In four Afro-American families [9, 10] and two Finnish families [11], Leu129Pro mutation in the GPIba gene has been identified. In several families the Ala156Val mutation has been identified [12]. In two families from South Iran, the Phe55Ser missense mutation has been identified in the GPIX gene [13]. Tyr88Cys mutation in the GPIBB gene has been identified in several Japanese families [14], in two Japanese and in one Saudi family. A 13-bp deletion of the signal peptide coding sequence leading to premature termination has been found [15, 16]. Ser stop in the GPIBB gene have been reported in three Tunisian families [17]. Ten novel mutations were identified in India. Out of these eight were frame shift mutations including p.Asp79GlufsX2, p.Phe314PhefsX37, p.Pro93ProfsX59, p.Asp89GlufsX63, p.Glu489AsnfsX64, p.Phe355PhefsX4, p.Leu479PhefsX19 and p.Leu531ArgfsX22, one missense mutation (p.Val262Gly) in GPIBA and one nonsense mutation (p.Tyr95X) in GP9. Also two other known mutations were identified including one missense mutation (p.Cys24Arg) in GP9 and one nonsense change (p.Trp46X) in GPIBB [18]. In our baby the whole exome sequencing identified the homozygous 6-bp duplication c.511_516dup p.(Leu171_Leu172dup) in exon 2 of GP1BB gene. To the best of knowledge this variant has neither been described in literature, nor been annotated in genetic data base. Family analysis revealed that the brother and father of the index patient also carry this variant in the homozygous state, while the mother is a heterozygous carrier. Bleeding symptoms may manifest after birth or during child hood. Presentations include purpura, gum bleeds, epistaxis and rarely gastrointestinal bleeding hematuria [1]. Our baby did not have any manifestation of bleeding in the neonatal period and in infancy on follow-up. There is thrombocytopenia, giant platelets in our case, her father and her brother; but there is no history of any bleeding.
Conclusions
This case report illustrates that peripheral smear’s platelets morphology should be evaluated whenever thrombocytopenia is diagnosed. This evaluation may give a clue to the correct diagnosis and can lead to the correct diagnosis as occurred in our patient. Whenever BSS is suspected proper family history should be obtained, as this can also give a clue to the diagnosis.
Acknowledgments
None to declare.
Financial Disclosure
No grants or any financial support from any individual or any company.
Conflict of Interest
There is no conflict of interest of any author
Informed Consent
The consent was taken from father of the baby for publication of case report.
Author Contributions
All Authors have taken part in preparing and reviewing the case report.
Data Availability
The authors declare that data supporting the findings of this study are available within the article.
| References | ▴Top |
- Lanza F. Bernard-Soulier syndrome (hemorrhagiparous thrombocytic dystrophy). Orphanet J Rare Dis. 2006;1:46.
doi pubmed - Qiao J, Davis AK, Morel-Kopp MC, Ward CM, Gardiner EE, Andrews RK. Low levels of CD9 coincidental with a novel nonsense mutation in glycoprotein Ibbeta in a patient with Bernard-Soulier syndrome. Ann Hematol. 2015;94(12):2069-2071.
doi pubmed - Andrews RK, Lopez JA, Berndt MC. Molecular mechanisms of platelet adhesion and activation. Int J Biochem Cell Biol. 1997;29(1):91-105.
doi - Miller JL, Lyle VA, Cunningham D. Mutation of leucine-57 to phenylalanine in a platelet glycoprotein Ib alpha leucine tandem repeat occurring in patients with an autosomal dominant variant of Bernard-Soulier disease. Blood. 1992;79(2):439-446.
doi pubmed - Gresele P, Falcinelli E, Bury L. Laboratory diagnosis of clinically relevant platelet function disorders. Int J Lab Hematol. 2018;40(Suppl 1):34-45.
doi pubmed - Savoia A, Pastore A, De Rocco D, Civaschi E, Di Stazio M, Bottega R, Melazzini F, et al. Clinical and genetic aspects of Bernard-Soulier syndrome: searching for genotype/phenotype correlations. Haematologica. 2011;96(3):417-423.
doi pubmed - Hadjkacem B, Elleuch H, Gargouri J, Gargouri A. Bernard-Soulier syndrome: novel nonsense mutation in GPIbbeta gene affecting GPIb-IX complex expression. Ann Hematol. 2009;88(5):465-472.
doi pubmed - Lopez JA, Andrews RK, Afshar-Kharghan V, Berndt MC. Bernard-Soulier syndrome. Blood. 1998;91(12):4397-4418.
doi pubmed - Antonucci JV, Martin ES, Hulick PJ, Joseph A, Martin SE. Bernard-Soulier syndrome: common ancestry in two African American families with the GP Ib alpha Leu129Pro mutation. Am J Hematol. 2000;65(2):141-148.
doi - Li C, Martin SE, Roth GJ. The genetic defect in two well-studied cases of Bernard-Soulier syndrome: a point mutation in the fifth leucine-rich repeat of platelet glycoprotein Ib alpha. Blood. 1995;86(10):3805-3814.
doi pubmed - Koskela S, Javela K, Jouppila J, Juvonen E, Nyblom O, Partanen J, Kekomaki R. Variant Bernard-Soulier syndrome due to homozygous Asn45Ser mutation in the platelet glycoprotein (GP) IX in seven patients of five unrelated Finnish families. Eur J Haematol. 1999;62(4):256-264.
doi pubmed - Savoia A, Balduini CL, Savino M, Noris P, Del Vecchio M, Perrotta S, Belletti S, et al. Autosomal dominant macrothrombocytopenia in Italy is most frequently a type of heterozygous Bernard-Soulier syndrome. Blood. 2001;97(5):1330-1335.
doi pubmed - Afrasiabi A, Lecchi A, Artoni A, Karimi M, Ashouri E, Peyvandi F, Mannucci PM. Genetic characterization of patients with Bernard-Soulier syndrome and their relatives from Southern Iran. Platelets. 2007;18(6):409-413.
doi pubmed - Kurokawa Y, Ishida F, Kamijo T, Kunishima S, Kenny D, Kitano K, Koike K. A missense mutation (Tyr88 to Cys) in the platelet membrane glycoprotein Ibbeta gene affects GPIb/IX complex expression—Bernard-Soulier syndrome in the homozygous form and giant platelets in the heterozygous form. Thromb Haemost. 2001;86(5):1249-1256.
doi pubmed - Watanabe R, Ishibashi T, Saitoh Y, Shichishima T, Maruyama Y, Enomoto Y, Handa M, et al. Bernard-soulier syndrome with a homozygous 13 base pair deletion in the signal peptide-coding region of the platelet glycoprotein Ib(beta) gene. Blood Coagul Fibrinolysis. 2003;14(4):387-394.
doi pubmed - Strassel C, Alessi MC, Juhan-Vague I, Cazenave JP, Lanza F. A 13 base pair deletion in the GPIbbeta gene in a second unrelated Bernard-Soulier family due to slipped mispairing between direct repeats. J Thromb Haemost. 2004;2(9):1663-1665.
doi pubmed - HadjKacem B, Elleuch H, Trigui R, Gargouri J, Gargouri AF. The same genetic defect in three Tunisian families with Bernard Soulier syndrome: a probable founder Stop mutation in GPIbbeta. Ann Hematol. 2010;89(1):75-81.
doi pubmed - Ali S, Ghosh K, Shetty S. Novel genetic abnormalities in Bernard-Soulier syndrome in India. Ann Hematol. 2014;93(3):381-384.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
International Journal of Clinical Pediatrics is published by Elmer Press Inc.