

| International Journal of Clinical Pediatrics, ISSN 1927-1255 print, 1927-1263 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, Int J Clin Pediatr and Elmer Press Inc |
| Journal website https://www.theijcp.org |
Case Report
Volume 11, Number 2, June 2022, pages 35-38

Pediatric Neuromyelitis Optica Spectrum Disorder Presenting as Intractable Vomiting
William Bakshta, Alex Ankara, Nikita Shuklaa, b
aDepartment of Child Neurology and Developmental Neurosciences, Texas Children’s Hospital/Baylor College of Medicine, Houston, TX, USA
bCorresponding Author: Nikita Malani Shukla, Department of Neurology, Texas Children’s Hospital/Baylor College of Medicine, Houston, TX 77005, USA
Manuscript submitted January 24, 2022, accepted April 6, 2022, published online June 16, 2022
Short title: A Case of Intractable Vomiting
doi: https://doi.org/10.14740/ijcp478
| Abstract | ▴Top |
Pediatric neuromyelitis optica spectrum disorder (NMOSD) is an autoimmune demyelinating disease that affects the central nervous system. Common presentations include weakness and sensory changes secondary to transverse myelitis, or vision loss due to optic neuritis. Area postrema syndrome, characterized by intractable nausea, vomiting, or hiccups secondary to a demyelinating lesion in the chemoreceptor trigger zone, is part of the core diagnostic criteria for NMOSD. However, it has infrequently been described as an initial presentation of pediatric NMOSD. Here, we present the case of a 13-year-old female who presented with intractable vomiting, which was initially presumed to be secondary to gastrointestinal pathology. It was not until she developed transverse myelitis that the etiology of her intractable vomiting was identified as area postrema syndrome secondary to NMOSD. She was successfully treated with rituximab, and she remains relapse-free after the initiation of immunosuppressive therapy. This case highlights this rare presentation of NMOSD and emphasizes the need for increased awareness amongst pediatric providers in recognizing this diagnosis and preventing delays in treatment.
Keywords: AQP4-IgG; NMOSD; MOG-IgG
| Introduction | ▴Top |
Neuromyelitis optica spectrum disorder (NMOSD) is a relapsing autoimmune demyelinating disease of the central nervous system (CNS) that primarily affects the spinal cord and the optic nerves [1]. Pediatric NMOSD, defined as disease presentation prior to 18 years of age, accounts for only 3-5% of all NMOSD cases [2]. Antibodies to the aquaporin-4 receptor (AQP4-IgG), which are located on the foot processes of astrocytes, have been identified as pathologic, and are part of the diagnostic criteria for NMOSD [1]. Approximately 65% of pediatric patients diagnosed with NMOSD are found to be seropositive for AQP4-IgG, though seropositivity may not occur until as late as 4 years after the initial attack [3, 4]. Serial testing is recommended in suspicious cases in which serum antibodies are not initially identified. Myelin oligodendrocyte glycoprotein (MOG), a glycoprotein expressed exclusively in oligodendrocytes, is implicated in approximately 40% of pediatric patients testing negative for AQP4-IgG [3]. Symptoms of NMOSD can include numbness and weakness of the extremities, and bowel and bladder difficulties secondary to transverse myelitis, or vision loss secondary to optic neuritis. Encephalopathy secondary to lesions in the cerebral hemispheres may also be observed. Less commonly, intractable nausea, vomiting, and hiccups can occur in the setting of area postrema syndrome, which is a demyelinating lesion affecting the chemoreceptor trigger zone of the medulla. Here, we present a case of pediatric NMOSD that was initially diagnosed as post-infectious gastroparesis with intractable vomiting.
| Case Report | ▴Top |
A 13-year-old previously healthy female presented to a freestanding emergency room (ER) in September 2019 with symptoms of acute onset nausea and vomiting. She had no past medical or surgical history and her family and environmental history were unremarkable. She was not on any medications. Her initial workup was unrevealing, and she was referred to pediatric gastroenterology (GI) for evaluation. She was seen in GI clinic the next month and endorsed approximately 4 weeks of persistent symptoms. She reported that symptoms began 2 days after eating a moldy orange. She then developed episodes of non-bloody, non-bilious emesis 4 - 5 times daily. Other symptoms included epigastric abdominal pain, dizziness with positional changes, and intermittent blurry vision. Lab workup revealed a mild leukopenia and mild anemia. A liver panel, lipase, and amylase were normal. She also had an abdominal X-ray which was normal. She was diagnosed with post-infectious gastroparesis and was prescribed omeprazole, as well as erythromycin for gastric motility. Given the concern for neurologic symptoms, neuroimaging was ordered by the GI provider. Magnetic resonance imaging (MRI) of the brain with contrast was obtained which was normal. The patient returned to GI clinic in November 2019, at which time her vomiting had improved, but she continued to have nausea. At this visit, she endorsed weakness in her lower extremities, for which a referral to pediatric neurology was placed. A clinic appointment was scheduled for February 2020, but the patient began having difficulty walking with frequent falls beginning in January 2020. Thus, she presented to an emergency center (EC) for further evaluation. Vital signs in the EC were normal. History obtained by the EC physician revealed symptoms of upper and lower extremity weakness, diminished sensation from the neck down, and urinary incontinence and constipation. Physical exam revealed weakness in all four extremities, diminished sensation below her neck, hyperreflexia, and clonus. She denied pain with eye movements, loss of color saturation, and had a normal ophthalmological exam. Routine labs including complete blood cell (CBC) and comprehensive metabolic panel were normal. MRI of the brain with contrast was obtained which showed concerns for bilateral optic neuritis (Fig. 1). MRI of the cervical and thoracic spine were obtained, which showed multiple T2 hyperintense lesions concerning for longitudinal transverse myelitis, one of which extended from the cranio-cervical junction to C6, and another from T8 through T11 (Fig. 2). She was admitted to the pediatric intensive care unit (PICU) given the risk of respiratory decompensation. She was treated with 5 days of high-dose intravenous steroids (30 mg/kg of intravenous methylprednisolone) followed by six sessions of plasmapheresis. She was then given 2 g/kg intravenous immunoglobulin (IVIg). She had significant improvement in her symptoms after treatment and was discharged home with outpatient therapies. Serum testing performed while inpatient showed positive AQP4-IgG with a titer of 1:1,000, confirming a diagnosis of seropositive NMOSD. Due to her diagnosis, her initial MRI performed in 2019 was reviewed once more, which was ultimately found to have increased T2 signal in the dorsal aspect of the medulla, consistent with area postrema involvement (Fig. 3). Therefore, this supports that her initial presentation of intractable vomiting several months prior was likely the first clinical signs of her NMOSD. After being diagnosed, she was started on rituximab infusions (1,000 mg for the first two doses, followed by 750 mg every 6 months), administered every 6 months, and has had no relapses since her initial hospitalization 2 years ago.
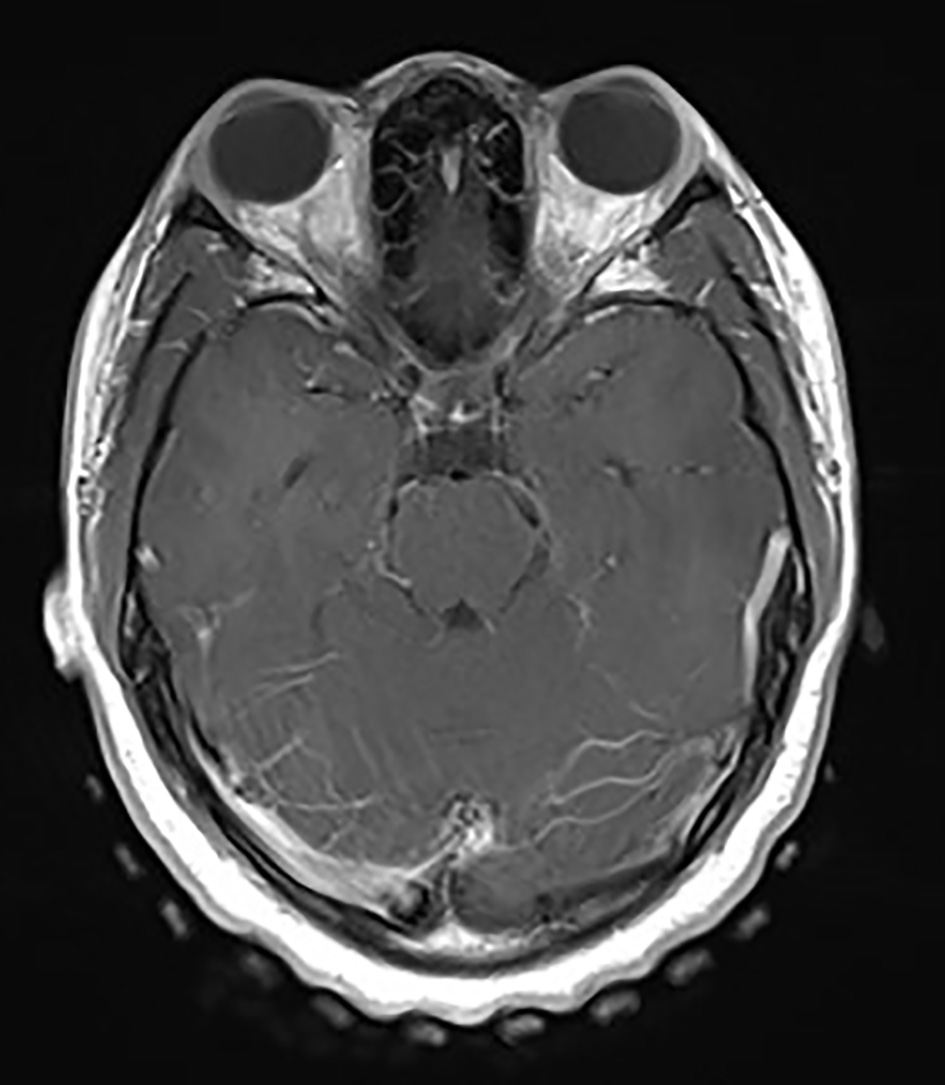 Click for large image | Figure 1. Contrast-enhanced axial T1 image showing bilateral optic neuritis. |
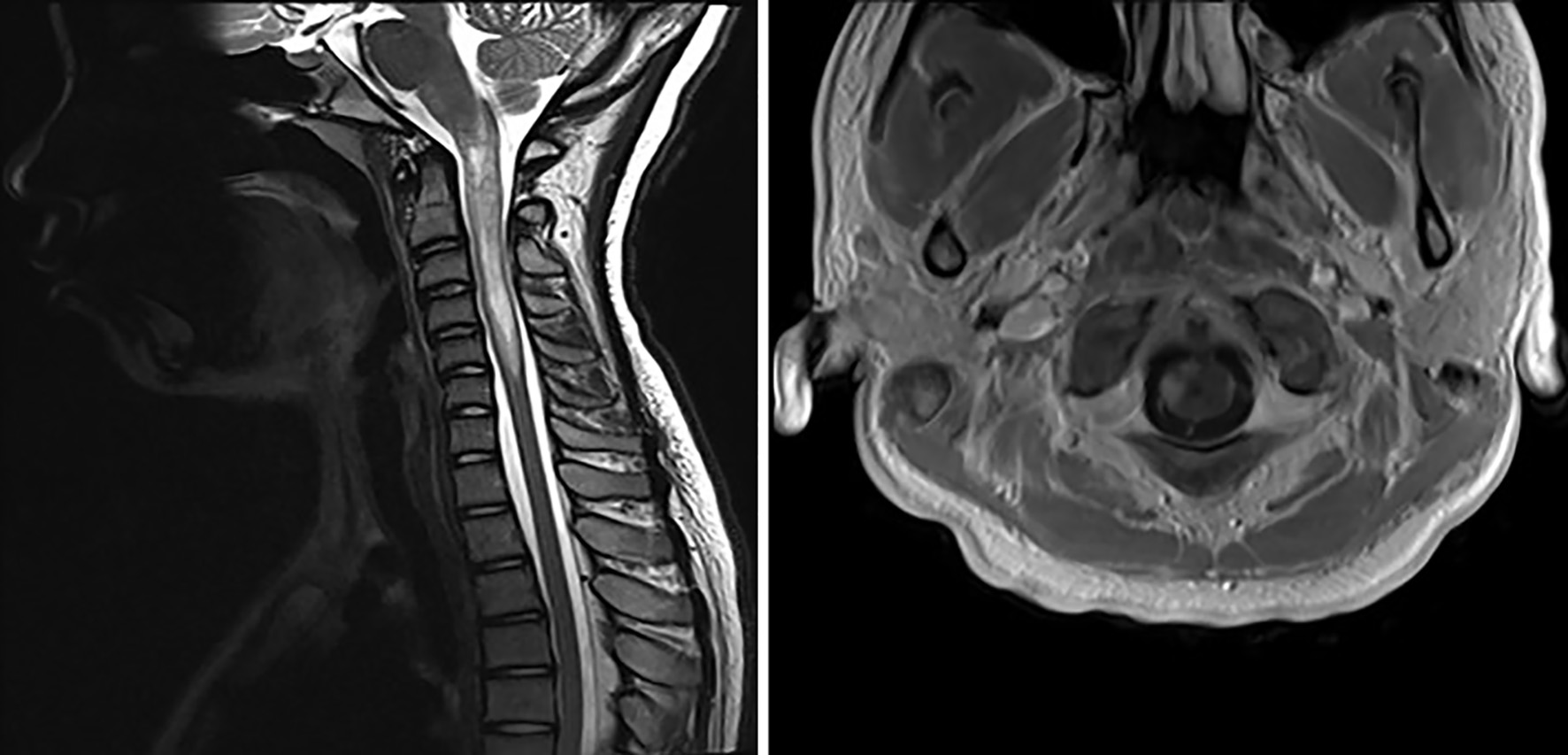 Click for large image | Figure 2. T2-weighted sagittal and axial MRI showing hyperintense lesions throughout the cervical and thoracic spine consistent with longitudinally extensive transverse myelitis. MRI: magnetic resonance imaging. |
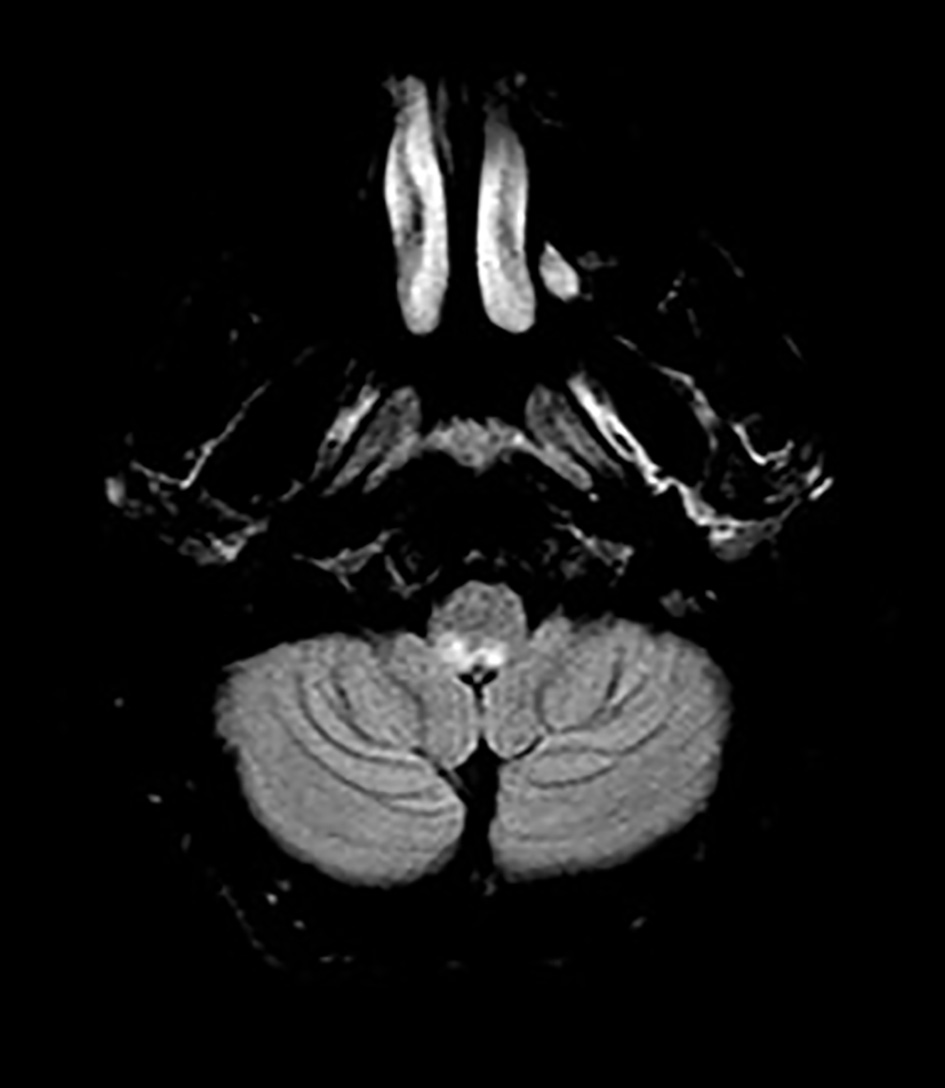 Click for large image | Figure 3. Initial MRI showing T2 hyperintensity in the dorsal medulla consistent with area postrema syndrome. MRI: magnetic resonance imaging. |
| Discussion | ▴Top |
Here, we report a case of pediatric onset NMOSD, initially presenting with intractable vomiting secondary to area postrema syndrome. She was ultimately both diagnosed and treated for her condition after developing more classic presenting symptoms, such as transverse myelitis and optic neuritis. Area postrema syndrome is a part of the diagnostic criteria for NMOSD but has less frequently been reported as a presenting symptom in pediatric NMOSD [1].
NMOSD is a relapsing demyelinating disease of the CNS that can affect the spinal cord, optic nerves, and less commonly the brain [1, 2]. Left untreated, NMOSD can lead to severe disability related to spinal cord lesions from transverse myelitis, or progressive vision loss related to optic neuritis [1]. Pediatric NMOSD accounts for 3-5% of all NMOSD [2]. The typical age of onset for pediatric disease is approximately 10 - 12 years, and there is a female predominance with a ratio of 9:1. Of patients, 73% are reported to be non-Caucasian, and 24% of those are reported to be African-American [2]. Pediatric NMOSD is thought to be more active than other pediatric demyelinating diseases, including pediatric multiple sclerosis, and these patients tend to accrue more disability within the first 2 years of diagnosis [4].
Based on the International Panel for NMO Diagnosis (IPND) 2015 criteria, the diagnosis of NMOSD involves the presence of one core criterion plus the presence of AQP4-IgG. Core criteria are broad and include syndromes of optic neuritis, acute myelitis, area postrema syndrome, acute brainstem syndrome, diencephalic syndrome, and symptomatic cerebral syndrome. If AQP4-IgG testing is negative, then the presence of two core clinical criteria with appropriate MRI findings is required for diagnosis [2]. Initial presentations of pediatric NMOSD can vary, but classic symptoms include a first clinical attack consisting of either optic neuritis, transverse myelitis, or both. Case series have shown that optic neuritis is a presenting symptom in approximately 50-75% of patients with pediatric NMOSD [1, 2]. This can present as partial or complete blindness in one or both eyes, as well as pain with extraocular movements [3]. Transverse myelitis has also been described as a first clinic attack in about 30-50% of pediatric NMOSD patients, either with or without optic neuritis [1, 2]. Transverse myelitis associated with NMOSD is typically longitudinal and can extend three or more vertebral segments [3]. These case series also show that brainstem involvement, such as area postrema syndrome, frequently develops, but this is usually associated with a relapse and not part of the first clinical attack [1]. However, more recent case series have shown that vomiting has been described as a presenting symptom in approximately 38% of pediatric cases [4].
The case presented describes a patient who developed intractable nausea and vomiting, as well as vague neurological symptoms, after reportedly eating a moldy orange. She was initially treated for post-infectious gastroparesis, but her GI practitioner astutely recognized the atypical neurological symptoms, including intermittent blurry vision and lightheadedness, and appropriately ordered brain imaging. Unfortunately, her MRI was read incorrectly, and it was not until she developed the more classic symptoms of NMOSD, including optic neuritis and transverse myelitis, that she was correctly diagnosed. However, this case does highlight an atypical presentation of pediatric NMOSD and stresses the importance of consideration of a demyelinating pathology in the differential diagnoses of a patient presenting with intractable nausea and vomiting that is otherwise not explained.
Acknowledgments
The authors would like to acknowledge the patient and family for allowing us to publish this case report to help further the understanding of neuromyelitis optica spectrum disorder.
Financial Disclosure
None to declare.
Conflict of Interest
William Baksht, Alex Ankar and Nikita Shukla have no conflict of interest to disclose.
Informed Consent
Informed consent was obtained.
Author Contributions
Mr. Baksht, Dr. Ankar and Dr. Shukla conceptualized the case report, drafted the initial manuscript, and reviewed and revised the manuscript. All authors approved the final manuscript as submitted and agree to be accountable for all aspects of the work.
Data Availability
The authors declare that data supporting the findings of this study are available within the article.
| References | ▴Top |
- Tenembaum S, Chitnis T, Nakashima I, Collongues N, McKeon A, Levy M, Rostasy K. Neuromyelitis optica spectrum disorders in children and adolescents. Neurology. 2016;87(9 Suppl 2):S59-66.
doi pubmed - Tenembaum S, Yeh EA, Guthy-Jackson Foundation International Clinical Consortium. Pediatric NMOSD: a review and position statement on approach to work-up and diagnosis. Front Pediatr. 2020;8:339.
doi pubmed - Chitnis T. Pediatric central nervous system demyelinating diseases. Continuum (Minneap Minn). 2019;25(3):793-814.
doi pubmed - Chitnis T, Ness J, Krupp L, Waubant E, Hunt T, Olsen CS, Rodriguez M, et al. Clinical features of neuromyelitis optica in children: US Network of Pediatric MS Centers report. Neurology. 2016;86(3):245-252.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
International Journal of Clinical Pediatrics is published by Elmer Press Inc.