

| International Journal of Clinical Pediatrics, ISSN 1927-1255 print, 1927-1263 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, Int J Clin Pediatr and Elmer Press Inc |
| Journal website http://www.theijcp.org |
Case Report
Volume 9, Number 2, June 2020, pages 59-63
Hypereosinophilia as a Presenting Symptom of Acute Lymphoblastic Leukemia in a Child
Boryana Avramovaa, b, Irena Vlahovaa, Atanas Bancheva, Konstantin Bachvarova, Denka Stoyanovaa, Dobrin Konstantinova
aDepartment of Pediatric Hematology and Oncology, University Hospital “Tzaritza Joanna”-ISUL, Sofia, Bulgaria
bCorresponding Author: Boryana Avramova, Department of Pediatric Hematology and Oncology, University Hospital “Tzaritza Joanna”-ISUL, 8 Bjalo More St, 1527 Sofia, Bulgaria
Manuscript submitted March 15, 2020, accepted April 2, 2020, published online June 18, 2020
Short title: Pediatric ALL With Hypereosinophilia
doi: https://doi.org/10.14740/ijcp365
| Abstract | ▴Top |
Hypereosinophilia is rare in children and represents primary or secondary conditions, the latter of which is associated with other diseases (parasitic infections and allergic, autoimmune, and neoplastic diseases). In small number of cases (< 1%) it is a part of the clinical presentation of acute lymphoblastic leukemia, the most common malignancy in children. More often hypereosinophilia precedes the symptoms of leukemia and hinders timely diagnosis, exposing patients in a high risk. In this report, a case of a child with acute lymphoblastic leukemia and hypereosinophilia is presented. The patient is a 30-month-old female, whose leukemia is preceded 2 weeks after hypereosinophilia. This symptom made the exact and timely diagnosis of oncological disease difficult. The further course of the disease was standard despite of delayed diagnosis and incomplete induction treatment due to the hepatic chemotoxicity. We present this case due to the rarity of hypereosinophilia in pediatric acute lymphoblastic leukemia. Every experience with such cases would be very useful for the improvement of the diagnosis of these patients. Moreover in our patient we did not find typical cytogenetic or molecular genetic abnormalities for this type of leukemia, and did not observe any clinical signs of organ complications specific for hypereosinophilia.
Keywords: Acute lymphoblastic leukemia; Hypereosinophilia; Children
| Introduction | ▴Top |
Hypereosinophilia (HE) is a relatively common finding in children, diagnosed when the absolute peripheral blood eosinophil count is over 500/µL. It is classified as a mild (500 - 1,500/µL), moderate (1,500 - 5,000/µL) and severe (> 5,000/µL). It may be primary or secondary (associated with other diseases), with the latest reports more often in children than in adults [1-4]. Parasitic infections, allergic reactions, immune deficiency, autoimmune and neoplastic diseases are most frequent causing secondary HE [2, 4], which by itself is due to T-cell dependent cytokine production [1, 3]. HE is more common in myeloid neoplasm in adult patients. In very rare cases (< 1%), especially in children, it is a part of the clinical presentation of acute lymphoblastic leukemia (ALL) [1].
ALL with eosinophilia was first described by Spitzer and Garson [5]. Until now, there are no more than 50 cases of HE with pediatric ALL reported in the literature. In most cases, HE precedes the occurrence of the ALL symptoms, and hinders the diagnosis, causing a risk to patients. Specific leukemic clone cytogenetic abnormalities are t(5;14) (q31:32) and del(5)(5q33). The prognosis of ALL with HE, both in children and adults, is critical [6].
We report a case of a female child with ALL presenting with HE. Due to the rarity of this symptom in pediatric ALL, every experience would be useful for the improvement of the diagnosis of these patients. Moreover, we did not find typical cytogenetic or molecular genetic abnormalities for this type of leukemia described in the literature, and did not observe any clinical signs of organ complications specific for HE (cardiac, renal, pulmonary, skin, central nervous system (CNS), etc.) [4-6].
| Case Report | ▴Top |
A 30-month-old female child was admitted to our department with a 2-week history of intermittent fever, mild itching and HE in the peripheral blood. In the child’s past medical history there is nothing to note, she had no past or chronic illness, surgery, or allergies. The child had all and timely immunizations according to the national immunization schedule. The parents reported a family history of lung sarcoidosis (mother) and hepatic steatosis (father).
The physical and neuropsychic development was normal, so was the growth (in 50th percentile for the length and 25th for the weight). Vital signs showed: temperature 38.8 °C (axillary), pulse 126 /min, respiratory rate 28/min, blood pressure (BP) 100/60 mm Hg. The patient was a well-developed, well-nourished toddler, in moderate damaged general state. Physical examination showed that head: normocephalic and atraumatic; eyes: both pupils are equal, round and reactive to light, no discharge, conjunctivitis or scleral icterus, no ptosis or periorbital edema; ears: both pinnae are in normal shape and contour, no preauricular pits or skin tags, no erythema or bulging; nose: normal pink mucosa, no discharge or blood visible; mouth: moist mucous membranes; pharynx: normal size of tonsils, no erythema or ulcerations; neck: grossly non-swollen; no lymphadenopathy, goiter or masses detected; chest: round chest cavity, no evidence of increased work of breathing; lungs: clear to auscultation bilaterally, no stridor, wheezes, crackles, or rubs; cardiovascular (CV) system: regular rate and rhythm of heart, no murmurs, gallops or rubs; normal pulses in all extremities; abdomen: distended, bowel signs present; liver edge was palpable 4 cm below costal margin; spleen: 5 cm below costal margin, no masses; extremities: warm, no cyanosis or edema, and no gross deformities; back: straight, no lordosis, no kyphosis; skin: pale, with hyperaemia on the face, without rash and hemorrhage, good skin turgor.
Neurological state showed that all extremities move symmetrically with appropriate tone. No clinical abnormalities were found with the CNS; symmetrical biceps, patellar and plantar reflexes, and negative Brudzinski’s and Kernig’s signs.
There was no clinical and laboratory evidence for parasitic infections or allergic diseases. We excluded organ infiltration and toxicity with chest X-ray, echocardiography and abdominal ultrasound.
From the laboratory tests (Table 1), hyperleukocytosis (white blood cell (WBC) 58,000/µL), HE (60%), and elevated levels of lactate dehydrogenase (LDH) (719 U/L) were registered. The levels of hemoglobin and platelet count were in normal range. The peripheral blood smear showed mature eosinophils, normal lymphocytes and neutrophils, and no blast cells. In cellular morphology of bone marrow aspiration we diagnosed > 50% lymphoid blasts cells (L1 type according to FAB classification), and eosinophil > 40% (Fig. 1). The bone marrow lymphoblast’s immunophenotype was cluster of differentiation 10 (CD10)+, CD19+, CD34+, CD45 low, CD20-, CD38+, and CD58+. The diagnosis was confirmed to be pre-B type ALL. Cytogenetic analysis of blast cells clone showed a normal 46 XX karyotype. Molecular genetic analysis was negative for BCR/ABL, TEL/AML1, MLL/AF4, E2A-Pbx1, CBfb-MYH11, and FLT3-ITD, and for the specific for HE translocations: FIP1L1-PDGFRA and FGFR.
 Click to view | Table 1. Results of Laboratory Test in Different Time Periods |
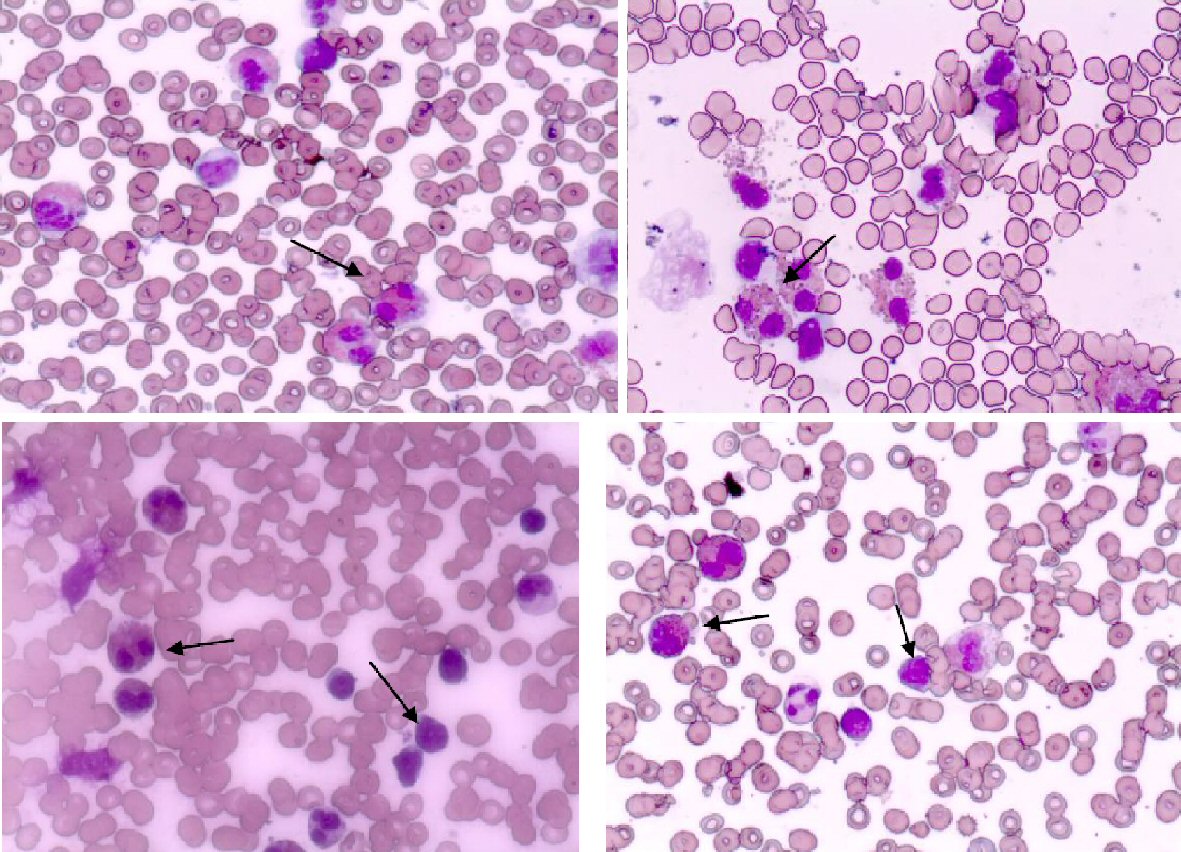 Click for large image | Figure 1. Peripheral blood with eosinophils and bone marrow aspiration smear with eosinophils and blast cells. |
The treatment was initiated according to the ALL IC BFM 2009 protocol [7]. On day 8 of induction phase of the treatment protocol, consisted of prednisone (60 mg/day) in a gradually increasing dose for 1 week, and one intrathecal application of methotrexate (10 mg), the patient had a good prednisone response (GPR), and on day 15 the bone marrow was classified as M1.
On day 22 (after the treatment with prednisone of 60 mg/day, one more intrathecal application of methotrexate of 10 mg, three applications of vincristine (1.5 mg weekly) and doxorubicin (30 mg weekly), and four intramuscular applications of L-asparaginase (10,000 U), a high-grade fever occurred and persisted. The blood cultures (three in three consecutive days) and urine culture were negative, but Candida albicans was isolated from faeces. From day 22 progressively increasing levels of aspartate aminotransferase (AST), alanine transaminase (ALT), gamma-glutamyl transferase (GGT), alkaline phosphatase (ALP), LDH, hypoproteinemia and hypoalbuminemia were registered. Bilirubin level and prothrombin time are also abnormal (Table 1). Subsequently, ascites and periorbital and lower limb edema developed.
The tests for viral (hepatitis A, B and C, herpes simplex, Epstein-Barr virus, cytomegalovirus, and parvovirus B19) infection were negative. Abdominal ultrasound showed no signs of venous occlusive disease, only hepatomegaly was detected. We registered severe hepatic drug toxicity with diffuse hepatic steatosis from the liver biopsy and stopped the chemotherapy.
After the symptomatic and supportive care treatment with Unasyn intravenous (IV) 2 × 1 g/day; vancomycin IV 4 × 150 mg/day; quamatel IV 10 mg/day; trans-methyl 2 × 150 mg/day IV; Ursofalk 125 mg/day per os (PO), 20% albumin 50 mL/day, Bactrim IV 3 × 500 mg/day for 10 days; and vitamin K IV 5 mg /week for 4 weeks; and erythrocyte, platelet and fresh frozen plasma (FFP) transfusions, a slow improvement in the laboratory tests results was seen over the following weeks, and the hepatic injury resolved completely within 1.5 months (Table 1).
The antileukemic treatment was restored. Further chemotherapy according to the protocol was well tolerated and the treatment was discontinued after 2 years. The patient, despite the expected poor prognosis of ALL with HE and the treatment delay, is currently in complete remission more than 3 years after the diagnosis.
| Discussion | ▴Top |
In this report, we present a pediatric case of HE as the first symptom of ALL, with clinical features of HE, but without specific initial laboratory findings of ALL, specific for this type of leukemia cytogenetic abnormalities, and organ toxicity from the HE.
ALL with HE is very rare in children and constitutes for less than 1% of all cases [1, 6]. In the medical literature till now approximately 50 patients with this constellation were reported [1, 8].
In most cases ALL with HE has a B-cell origin, and is more common in males patients. Very often the disease was presented with non-specific symptoms, such as fever, fatigue, respiratory symptoms, skin erythema and CV alterations. An increased risk of thromboembolic events was also found [1]. In a 10-year period, 222 children with ALL were treated in our department and this was the first case presented with HE at diagnosis. Most of described cases were with similar clinical presentation and laboratories findings of the disease; and ALL is from the B-cell lineage, as that in our patient [1, 6, 9].
In most cases HE precedes the occurrence of ALL by a long time (1 - 9 months), and disappears after the initiation of therapy or in remission. Very often at the onset of the disease there are no blast cells in the peripheral blood [1, 3, 4, 6, 9]. In our case we did not find peripheral blood blast cells at the initial presentation of HE, but confirmed the diagnosis very quickly after the bone marrow aspiration. The HE disappeared after the first week of chemotherapy.
Parasole et al have described two pediatric cases with HE in ALL, and suggested that the eosinophilia is a reactive condition to some pathogens, such as seasonal infection [6]. Other investigators also have demonstrated the non-clonal origin of eosinophils in ALL [5, 10, 11], but remarked that further cytogenetic and molecular studies are required to confirm these observations, given the rarity of this association. The most common described cytogenetic abnormalities in this type of ALL are t(5;14) (q31;q32) and del(5) (q15q33). Because chromosome 5 carries IL-3 gene, its overexpression leads to increased eosinophil production [6, 12]. Most authors found cytogenetic abnormalities only in leukemic blast cells and not in the eosinophils [6]. In our case we did not find typical cytogenetic or molecular genetic abnormalities of t(5;14) (q13;q32), del 5 (q15;q33) and PDGFRA, PDGFRB, FGFR1, and PCM1-JAK2.
In most of the described patients with HE and ALL, a significantly increased risk of organ complications from the eosinophil infiltration (cardiac, vascular thrombosis, pruritic erythroderma, lung infiltration and CNS toxicity) exists [8, 9, 14]. Congestive heart failure is the main cause of death in patients with ALL and HE [8, 13]. Parasole et al observed a thrombotic complication at diagnosis in their patient, which is successfully resolved with anticoagulant treatment [6]. Other authors reported pruritic erythroderma as an additional common presenting feature in children with ALL and HE [8, 9, 13]. In our patient we did not observe any clinical signs of complications specific for HE (cardiac, vascular, renal, pulmonary, CNS and other organ systems), except a light face hyperaemia. Maybe this is due to the quick diagnosis. But our patient has developed severe drug-induced hepatic toxicity from the antileukemic chemotherapy. No risk factors were observed in the patient, and the clinical manifestation of liver injury was sudden and severe, but transient with full recovery. Therefore, individual sensitivity to the combined treatment with L-asparaginase and steroids, rather than HE complication, should be discussed.
According to many authors early diagnosis of ALL is very important for adequate and successful treatment [1, 5, 6]. But in most cases HE precedes signs and symptoms of ALL by several weeks or months, and the onset is different from the usual one in ALL, with signs of eosinophil infiltration of body organs [5, 6]. Diagnosis can be difficult and delayed with high risk for the patient [1]. In our case we diagnosed ALL very quickly, although the lack of peripheral blood blast cell, following the existing algorithm for the differential diagnosis of HE; and this gave us a possibility to start the treatment of oncological disease on time [4].
HE in ALL is considered to be a poor prognostic marker both in children and adults [6], with median survival of 7.5 months [8], because of organ infiltration and related life-threatening complications [1, 6, 14, 15]. In comparison to standard ALL, congestive heart failure is the main cause of increased mortality in these patients [8]. Our patient is alive and in remission more than 3 years, without any complications.
Conclusions
As in our case and other reported in the literature, ALL is one of the possible causes of HE in children. Despite the rarity among all cases of HE, malignant disease, including ALL, should be suspected even in the absence of blasts cells in the peripheral blood. This is imperative for the timely diagnosis, treatment and improved prognosis of the disease. Due to the negative prognosis of this type of ALL in most cases of children and adults, and the increased risk of complications, bone marrow aspiration and biopsy are mandatory for all cases of secondary HE.
Acknowledgments
The authors would like to thank all the physician and nurses who participated in the diagnosis and treatment of the patient.
Financial Disclosure
Authors have no financial relationships relevant to this article to disclose.
Conflict of Interest
The authors have no conflict of interest to disclose.
Informed Consent
Informed consent for publication of this report was obtained from the parents of the patients.
Author Contributions
B. Avramova and I. Vlahova conceptualized and designed the case, drafted the initial manuscript, and reviewed and revised the manuscript. A. Banchev, D. Stoyanova and K. Bachvarov collected data, carried out the initial description, and reviewed and revised the manuscript. D. Konstantinov conceptualized and designed the case, had substantial contribution in the acquisition, analysis and interpretation of data, in the drafting of the article and revised it critically for important intellectual content. All authors approved the final manuscript as submitted and agree to be accountable for all aspects of the work.
Data Availability
The authors declare that data supporting the findings of this study are available within the article.
| References | ▴Top |
- Ferruzzi V, Santi E, Gurdo G, Arcioni F, Caniglia M, Esposito S. Acute lymphoblastic leukemia with hypereosinophilia in a child: case report and literature review. Int J Environ Res Public Health. 2018;15(6):1169.
doi pubmed - Yamaoka M, Akiyama M, Ohyama W, Tachimoto H, Matsumoto K. Acute lymphoblastic leukemia with hypereosinophilia in a 3-year-old boy. Pediatr Int. 2018;60(1):88-90.
doi pubmed - Ali AM, Rashidi A, DiPersio JF. Acute lymphoblastic leukemia presenting with hypereosinophilia: Case report and review of the literature. Blood Cells Mol Dis. 2017;65:97-100.
doi pubmed - Amshalom A, Lev A, Trakhtenbrot L, Golan H, Weiss B, Amariglio N, Rechavi G, et al. Severe eosinophilia in children: a diagnostic dilemma. J Pediatr Hematol Oncol. 2013;35(4):303-306.
doi pubmed - Spitzer G, Garson OM. Lymphoblastic leukemia with marked eosinophilia: a report of two cases. Blood. 1973;42(3):377-384.
doi pubmed - Parasole R, Petruzziello F, De Matteo A, Maisto G, Castelli L, Errico ME, Menna G, et al. Hypereosinophilia in childhood acute lymphoblastic leukaemia at diagnosis: report of 2 cases and review of the literature. Ital J Pediatr. 2014;40:36.
doi pubmed - Natch U, Chattopadhyay D, Bakliwal A, Vaniyath S, Chetia R, Rajoreya AK. Encouraging results with ALL IC-BFM 2009 protocol therapy in pediatric acute lymphoblastic leukemia/lymphoma in resource-limited setting: a single-center study from India. Blood. 2019;134(Suppl 1):5118.
doi - Rezamand A, Ghorashi Z, Ghorashi S, Nezami N. Eosinophilic presentation of acute lymphoblastic leukemia. Am J Case Rep. 2013;14:143-146.
doi pubmed - Ayhan AC, Timur C, Ayhan Y, Cakir B, Erguven M. Does hypereosinophilic syndrome precede common B acute lymphoblastic leukaemia in childhood? A case report. Acta Haematol. 2012;127(2):90-92.
doi pubmed - Bhatti FA, Hussain I, Ali MZ. Adult B lymphoblastic leukaemia/lymphoma with hypodiploidy (-9) and a novel chromosomal translocation t(7;12)(q22;p13) presenting with severe eosinophilia - case report and review of literature. J Hematol Oncol. 2009;2:26.
doi pubmed - Wynn TT, Heerema NA, Hammond S, Ranalli M, Kahwash SB. Acute lymphoblastic leukemia with hypereosinophilia: report of a case with 5q deletion and review of the literature. Pediatr Dev Pathol. 2003;6(6):558-563.
doi pubmed - Grimaldi JC, Meeker TC. The t(5;14) chromosomal translocation in a case of acute lymphocytic leukemia joins the interleukin-3 gene to the immunoglobulin heavy chain gene. Blood. 1989;73(8):2081-2085.
doi - Williams E, Smart SC, Go RS. Catastrophic thromboembolism in a patient with acute lymphoblastic leukemia and hypereosinophilia. Haematologica. 2004;89:e15.
- Files MD, Zenel JA, Armsby LB, Langley SM. A child with eosinophilia, Loeffler endocarditis, and acute lymphoblastic leukemia. Pediatr Cardiol. 2009;30(4):530-532.
doi pubmed - Girodon F, Bergoin E, Favre B, Martha SA, Mugneret F, Couillault G, Carli PM, et al. Hypereosinophilia in acute B-lineage lymphoblastic leukaemia. Br J Haematol. 2005;129(5):568.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
International Journal of Clinical Pediatrics is published by Elmer Press Inc.