

| International Journal of Clinical Pediatrics, ISSN 1927-1255 print, 1927-1263 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, Int J Clin Pediatr and Elmer Press Inc |
| Journal website http://www.theijcp.org |
Case Report
Volume 5, Number 3-4, December 2016, pages 47-50
Congenital Embryonal Rhabdomyosarcoma Presenting as a Cutaneous Nodule in a Neonate
Vishal Vishnu Tewaria, d, Ritu Mehtab, Kunal Tewaric
aDepartment of Pediatrics, Army Hospital (Referral & Research), New Delhi, India
bDepartment of Pathology, All India Institute of Medical Sciences, New Delhi, India
cDepartment of Anaesthesia, Base Hospital, New Delhi, India
dCorresponding Author: Vishal Vishnu Tewari, Department of Pediatrics, Army Hospital (Referral & Research), New Delhi 110010, India
Manuscript accepted for publication July 06, 2016
Short title: Congenital Cutaneous Rhabdomyosarcoma
doi: https://doi.org/10.14740/ijcp254e
| Abstract | ▴Top |
Rhabdomyosarcoma is the commonest malignant soft tissue sarcoma affecting children. It originates from the embryonic mesenchyme precursor of striated muscle and involves the head and neck region as a solitary or multiple skin lesion. A 20-day-old female neonate presented with a cutaneous nodule over left shoulder. Hematoxylin and eosin staining on the biopsy specimen showed malignant small round cell tumor. Immunohistochemistry studies were positive for myogenin (nuclear positivity) and desmin (cytoplasmic positivity). Presentation in the neonatal period is rare and poses a diagnostic and therapeutic challenge. The management involves complete excision of the tumor followed by chemotherapy.
Keywords: Congenital cutaneous rhabdomyosarcoma; Desmin; Embryonal cell; Immunohistochemistry; Myogenin
| Introduction | ▴Top |
Rhabdomyosarcoma (RMS) is a malignant mesenchymal tumor with striated muscle differentiation. RMS belongs to the class of small round blue cell tumors and has been classified on histological basis into embryonal, botryoid, spindle cell variant, alveolar and undifferentiated. It is commoner in young children with a peak age of occurrence at 4 years [1]. Only 6% of all RMS cases occur in infancy. It is extremely rare in neonates with the reported incidence in neonates of about 7.2 per 100,000 live births [2]. RMS arising before the age of 1 month is termed as congenital RMS. Congenital RMS represented 0.4% of all RMS cases in Intergroup Rhabdomyosarcoma Study Group (IRSG) [3]. It primarily involves the head and neck region but may also occur at other sites such as genitourinary tract, extremities and trunk [4]. Rarely RMS presents as a cutaneous nodular lesion with very scant literature available on this topic [3, 5]. A rare presentation of embryonal RMS masquerading as a congenital cutaneous nodule in 20 days old neonate is highlighted in this report.
| Case Report | ▴Top |
A 20-day-old female neonate presented with cutaneous nodular lesion overlying the left deltoid region. The swelling was present since birth and was progressively increasing in size. She was a full term normal delivery with a birth weight of 2.8 kg. Antenatal, intrapartum and postnatal periods were uneventful. There was no history suggestive of birth trauma. Maternal TORCH titer profile was negative for both IgG and IgM. The neonate had received the Bacillus Calmette-Guerin (BCG) vaccine intradermally on day 2 of age administered at the insertion of the deltoid on the left side. General physical and systemic examination was unremarkable. She was exclusively breast fed and she had gained 200 g weight since birth showing her to be thriving. The papulonodular swelling measured 2 × 2 × 1.5 cm in size and was ecchymotic, firm to hard, non-pedunculated, non-tender and was attached to the skin but was freely mobile from deeper tissues (Fig. 1). There was no organomegaly or lymphadenopathy. Her hematological and biochemical investigations were done which revealed Hb of 14.5 g/dL, TLC of 8,400 mm3, platelets of 165,000 mm3, BUN of 8 mg/dL, serum creatinine of 0.5 mg/dL, and total bilirubin of 1.4 mg/dL which were within normal limits. Urine analysis was also normal. Her chest X-ray showed a small soft tissue shadow but did not show any underlying bony growth and ultrasound evaluation of the lesion and abdomen was unremarkable. Examination of the fundus and cerebrospinal fluid (CSF) cytology was unremarkable. Contrast-enhanced CT (CECT) of chest and brain did not show any pulmonary lesions or parameningeal lesions. The neonate underwent a skin biopsy with a clinical diagnosis of hemangioma. Hematoxylin and eosin (H&E) stained sections from skin biopsy showed malignant small round cell tumor. The neoplastic cells had eosinophilic cytoplasm, round nuclei, vesicular chromatin and prominent nucleoli. The cells were infiltrating the dermis and the subcutaneous tissue. Rhabdomyoblasts were also present (Fig. 2a, b). Immunohistochemistry (IHC) panel was put up to further characterize the neoplastic cells. The cells were positive for myogenin (nuclear positivity) and desmin (cytoplasmic positivity) (Fig. 2c, d), while stains for leucocyte common antigen synaptophysin were negative. Based on histopathological and IHC findings, the neonate was diagnosed to have a congenital cutaneous embryonal RMS, stage 2 as per the IRSG staging system. The infant underwent complete resection of the tumor and was classified as group I as per IRSG surgical-pathologic grouping. This was followed by chemotherapy with VAC regimen comprising of vincristine, dactinomycin and cyclophosphamide. Vincristine was given as 1.5 mg/m2 intravenous (IV), cyclophosphamide at 2.2 g/m2 IV and dactinomycin at 0.015 mg/kg for 22 weeks. Subcutaneous granulocyte colony stimulating factor (G-CSF) for WBC recovery and IV mesna for uroprotection was given concurrently with cyclophosphamide. The infant tolerated the surgery and chemotherapy and has made uneventful recovery. She has been on regular follow-up and is showing normal growth and attainment of developmental milestones.
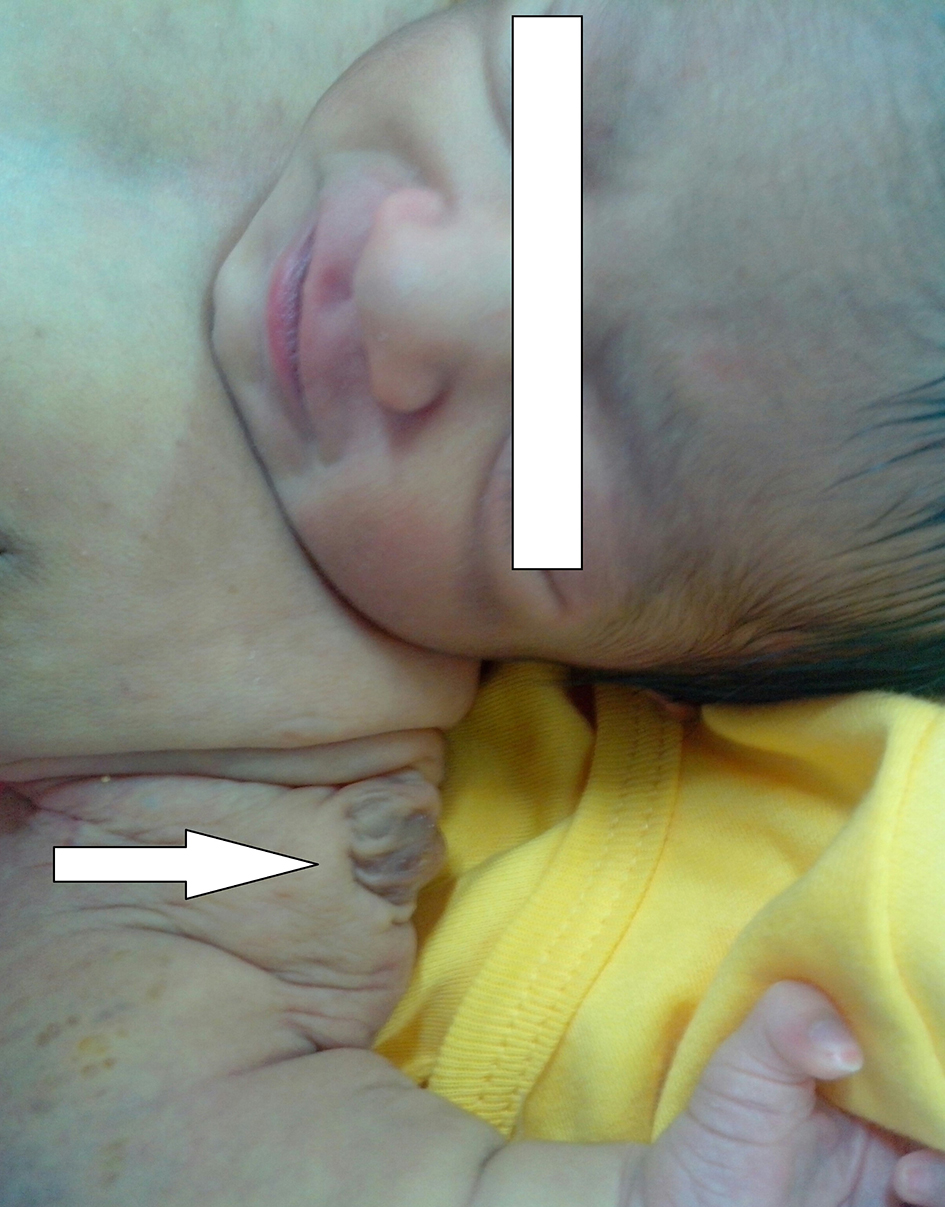 Click for large image | Figure 1. Photograph of the child showing papulonodular swelling over the left shoulder (white arrow). |
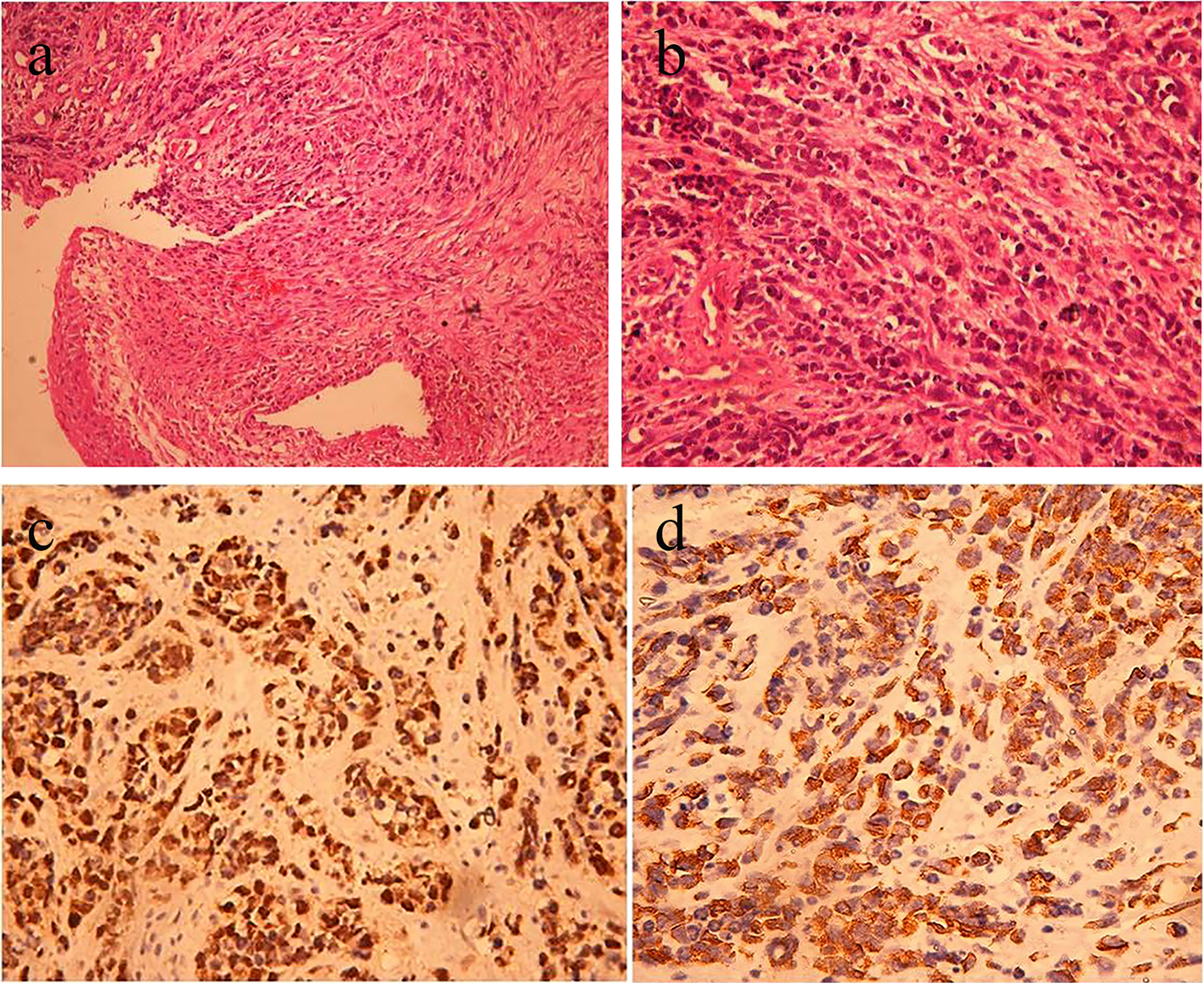 Click for large image | Figure 2. (a) H&E stained section from the cutaneous nodule showing infiltration of dermis and subcutaneous tissue by small round blue cells. The cells exhibit eosinophilic cytoplasm, round nuclei, vesicular chromatin and prominent nucleoli (magnification at × 100). (b) H&E stained section from the cutaneous nodule (magnification at × 400). (c) Immunohistochemistry showing nuclear positivity with myogenin. (d) Immunohistochemistry showing cytoplasmic positivity with desmin. |
| Discussion | ▴Top |
RMS is a malignant mesenchymal tumor of children first described by Zenker in 1856 [6]. It is the commonest soft tissue sarcoma in children and is the third commonest extracranial soft tissue tumor affecting children. It has an incidence in children of 4.5%. It also affects adults but incidence is much less as compared to the children, accounting for < 1% of all malignancies [7]. RMS shows bimodal distribution. It usually involves children below the age of 10 years with peak incidence reported by the age of 4 years. It is extremely rare in neonates and presentation at birth reflects intrauterine origin. About 0.4-1% of cases of RMS are congenital and it shows a slight male preponderance. Ferrari et al in an Italian co-operative study reporting on 50 infants with RMS over a 20-year period showed 15 cases to have congenital RMS [8]. The inherited syndromes associated with RMS are Li-Fraumeni syndrome, neurofibromatosis type 1 (NF1), Beckwith-Wiedemann syndrome, Costello syndrome and Noonan syndrome. It occurs predominantly in the head and neck region with orbit, paranasal sinuses and nasopharynx being the frequently involved sites. RMS in this region accounts for 30% of all head and neck region malignancies. Other sites of occurrence are genitourinary tract, extremities and retroperitoneum. Signs and symptoms of RMS depend upon the sites involved [9]. Depending upon the sites of involvement, various presentations include proptosis in orbital RMS, bleeding from nose or throat or swelling in head and neck region [10, 11]. Children with genitourinary RMS present with bowel and bladder disturbances along with hematuria while vaginal RMS commonly presents as a protruding polypoidal mass. Presentation as congenital cutaneous RMS is very rare with very few cases reported in literature. Our patient had presented in the neonatal period with a cutaneous nodule which was present since birth with an apparent increase in size. The diagnosis of RMS was made primarily on the basis of biospy and immunohistochemical examination along with radiological correlation. On histology striated muscle differentiation is commonly seen and is a clue towards diagnosis of RMS. Based on the International Classification of RMS, created by IRSG, four histological subtypes are identified. These are embryonal, alveolar, botryoid and spindle cell, and undifferentiated RMS [12]. Embryonal RMS is the commonest and it usually affects neonates and younger children. Alveolar RMS affects older individuals. Spindle cell RMS occurs primarily in the paratesticular region in children and carries a good prognosis, whereas in adults, spindle cell variant involves the head and neck region and is more aggressive. Spindle cell and botryoid carries superior prognosis, embryonal RMS carries intermediate prognosis and alveolar and undifferentiated RMS carries poor prognosis. Apart from histology, other prognostic markers include site of the lesion, size of the tumor and extent, lymph node status and metastasis. Irrespective of histology, RMS affecting limbs are more chemo-insensitive. However, Lobe et al showed that tumor histology, tumor size and type of surgery did not predict clinical outcome, while presence of necrosis and small round cell configuration coincides with a poor prognosis regardless of histological diagnosis [13]. In these patients, the 5-year survival is 55-70%. The optimal management strategy depends mainly on the stage and type of RMS with a multidisciplinary approach. Treatment modalities include surgical removal of the tumor followed by chemotherapy and radiotherapy. Complete resection of the tumor with safe margins is mandatory. IRS-IV data show local failure rates from extremities to be 7% for group III disease. Embryonal RMS responds very well to chemotherapy with the VAC regime [14]. Data from the IRS studies have shown that the addition of other chemotherapeutic agents like doxorubicin or etoposide is not required in group I disease and is of no benefit in group III or IV disease [15]. RMS is moderately radiation sensitive and thus higher doses of radiation are required. The use of radiotherapy is restricted due to its potential side effects in neonates and younger children. It is not indicated in group I stage 1 or 2, but is indicated for group I stage 3 and group II, III and IV disease [12]. This report illustrates a rare presentation of congenital embryonal RMS masquerading as a cutaneous nodule in a neonate. A high index of suspicion is needed on the part of the clinician along with skin biopsy for histopathology and immunohistochemical studies to confirm the diagnosis, correctly identify the histological subtype and to differentiate RMS from the other small round blue cell tumors like lymphoma, neuroblastoma and primitive neuroectodermal tumor. Management strategy should include complete excision of the nodule followed by chemotherapy with VAC regime.
Consent
Written informed consent was obtained from the patient for publication of this case report and accompanying images.
Conflicts of Interest
The authors declare no conflicts of interest.
Financial Disclosure
The authors declare no source of financial support.
| References | ▴Top |
- Wexler LH, Crist WM, Helman LJ, et al. Rhabdomyosarcoma and Undifferentiated sarcoma. In: Pizzo PA, Poplack DG, eds. Principles and Practice of Pediatric Oncology, 4th edn. Philadelphia: Lippincott Willams and Wilkins, 2002; 939-971.
- Ragab AH, Heyn R, Tefft M, Hays DN, Newton WA, Jr., Beltangady M. Infants younger than 1 year of age with rhabdomyosarcoma. Cancer. 1986;58(12):2606-2610.
doi - Brecher AR, Reyes-Mugica M, Kamino H, Chang MW. Congenital primary cutaneous rhabdomyosarcoma in a neonate. Pediatr Dermatol. 2003;20(4):335-338.
doi pubmed - Singh O, Gupta SS, Upadhyaya V, Sharma SS, Lahoti BK, Mathur KR. Rhabdomyosarcoma of the posterior chest wall in a newborn: a case report. Cases J. 2009;2:6818.
doi pubmed - Godambe SV, Rawal J. Blueberry muffin rash as a presentation of alveolar cell rhabdomyosarcoma in a neonate. Acta Paediatr. 2000;89(1):115-117.
doi - Stuart A, Radhakrishnan J. Rhabdomyosarcoma. Indian J Pediatr. 2004;71(4):331-337.
doi pubmed - Arul AS, Verma S, Verma R. Oral rhabdomyosarcoma-embryonal subtype in an adult: A rarity. J Nat Sci Biol Med. 2014;5(1):222-225.
doi pubmed - Ferrari A, Casanova M, Bisogno G, Zanetti I, Cecchetto G, De Bernardi B, Riccardi R, et al. Rhabdomyosarcoma in infants younger than one year old: a report from the Italian Cooperative Group. Cancer. 2003;97(10):2597-2604.
doi pubmed - Gura A, Tezcan G, Karaguzel G, Cevikol C, Oygur N. An unusual localization of embryonal rhabdomyosarcoma in a neonate. Turk J Pediatr. 2007;49(1):82-84.
pubmed - Singh GB, Arora R, Kumar D, Jain M, Puri V. A rare case of congenital rhabdomyosarcoma with review of the literature. Case Rep Otolaryngol. 2013;2013:518952.
doi - Vankalakunti M, Das A, Rao NK. Postauricular congenital alveolar rhabdomyosarcoma- a case report of an unusual entity. Diagn Pathol. 2006;1:37.
doi pubmed - Lanzkowsky P. In: Manual of Pediatric Hematology and Oncology 4th edn. Burlington, Mass: Elsevier Academic Press, 2005;561-584.
doi - Lobe TE, Wiener ES, Hays DM, Lawrence WH, Andrassy RJ, Johnston J, Wharam M, et al. Neonatal rhabdomyosarcoma: the IRS experience. J Pediatr Surg. 1994;29(8):1167-1170.
doi - Baker KS, Anderson JR, Link MP, Grier HE, Qualman SJ, Maurer HM, Breneman JC, et al. Benefit of intensified therapy for patients with local or regional embryonal rhabdomyosarcoma: results from the Intergroup Rhabdomyosarcoma Study IV. J Clin Oncol. 2000;18(12):2427-2434.
pubmed - Raney RB, Maurer HM, Anderson JR, Andrassy RJ, Donaldson SS, Qualman SJ, Wharam MD, et al. The Intergroup Rhabdomyosarcoma Study Group (IRSG): Major Lessons From the IRS-I Through IRS-IV Studies as Background for the Current IRS-V Treatment Protocols. Sarcoma. 2001;5(1):9-15.
doi pubmed
This is an open-access article distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
International Journal of Clinical Pediatrics is published by Elmer Press Inc.